
Activation of extracellular signal-regulated kinase during silibinin-protected, isoproterenol-induced apoptosis in rat cardiac myocytes is tyrosine kinase pathway-mediated and protein kinase C-dependent
Introduction
Heart failure remains one of the leading causes of death in all industrialized nations, and the quest for novel treatment options has recently directed attention to cardiac myocyte injury as a promising target, since this approach might provide the opportunity to prevent disease progression rather than treating established cardiomyopathy and heart failure[1,2]. It has been recently shown that beta-adrenergic agonists can induce apoptosis in cultured neonatal cardiac myocytes[3,4], suggesting that it might be one of the factors involved in myocardial cell injury in heart failure in vivo.
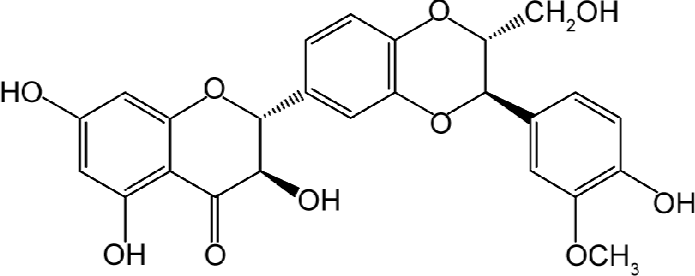
Silibinin (Figure 1), a polyphenolic flavanoid, exhibits potent antioxidant activity[5], in addition to hepatoprotec-tive[6,7] and anti-inflammatory effects[8]. Besides these, silibinin was found to have cytoprotective effects[9]. Numerous studies show that ageing is associated with increased rates of stress-induced apoptosis[10], and the cumulative effects of cell loss have been implicated in various diseases including neurodegeneration, retinal degeneration, cardiovascular disease and frailty[11–13]. Therefore, searching for the active compounds from Chinese herbal medicines which inhibit disease-associated apoptosis or protect cells from various causes of death is the aim of our study.
Previous studies pertaining to the components of the signaling system in cardioprotection have implicated a role for tyrosine kinases[14]. It has been increasingly apparent that G proteins are involved in the regulation of cell growth and differentiation as well as receptor protein tyrosine kinases (RPTK)[15]. At the intracellular face of the cell membrane, the exchange of GTP to GDP increases the affinity of the small G protein Ras for c-Raf, which then translocates to the membrane[16,17]. In RPTK-linked signaling, adaptor proteins such as Shc and growth factor receptor bound 2 (Grb2) interact directly with the phophorylated receptors to promote activation of Ras and Raf. Grb2 binds to the phosphotyosine through its SH2 domains and constitutively associates with polyproline domains in the GEF Sos. Binding of Grb2 to an RPTK thus places Sos in the vicinity of Ras GDP[18–20].
A mediator of G-protein-signaling that has become the focus of recent investigations is the protein kinase C (PKC) family which is comprised of at least 12 members that share structural homology and induce a great variety of intracellular responses when activated by lipid products of phospholipase C or D activity[21,22].
Biochemical and genetic studies in various cell systems have demonstrated that Raf-1 functions downstream of activated tyrosine kinases and Ras, and upstream of MAPK kinase (MEK) and mitogen-activated protein kinase (MAPK)[23,24]. Growing evidence suggests that modulation of complex network of MAPK signaling cascades could be a rewarding approach to treat cardiomyocyte hypertrophy and heart failure[25]. The MAPK are elements of 3-tiered protein kinase cascades and basically comprise 3 subfamilies: the extracellularly responsive kinases (ERK), the c-Jun N-terminal kinases (JNK), and the p38 MAPK. While the ERK are particularly implicated in growth-associated responses, the latter 2 groups are generally activated by cytotoxic stress factors[26].
In the present study, we demonstrate whether the tyrosine kinase pathway participates in silibinin-protected rat cardiac myocyte apoptosis, and whether the activity of upstream PKC is required for the increase of Ras and Raf-1. Among the downstream targets Ras and Raf-1 are of MAPK members. We tried to examine the expression of MAPK in a beta-adrenergic agonist, isoproterenol, in stimulation-induced injury of cultured rat neonatal cardiac myocytes after silibinin treatment.
Materials and methods
Chemical reagents Silibinin (Lot 0856–9902) was obtained from the Beijing Institute of Biologic Products (Beijing, China). The purity of silibinin was measured by HPLC and was determined to be about 99%. Silibinin was dissolved in DMSO to make a stock solution. The DMSO concentration was kept below 0.1% in all the cell cultures and did not exert any detectable effects on cell growth or cell death.
Genistein (a PTK inhibitor), manumycin A (a Ras inhibitor), GW5074 (a Raf-1 inhibitor) and staurosporine (a PKC inhibitor) were purchased from Enzyme Systems (Livermore, CA, USA). PD98059 (an ERK inhibitor), SP600125 (a JNK inhibitor) and SB203580 (a p38 MAPK inhibitor) were purchased from Calbiochem (La Jolla, CA, USA).
Fetal bovine serum (FBS) was from Tianjin TBD (Tianjin, China). Dulbecco’s modified Eagle’s medium (DMEM) was from Hyclone (Logan, UT, USA). Murine polyclonal antibodies against p-ERK, p-JNK and p-p38, rabbit polyclonal antibodies against Ras, Raf-1, Grb2, ERK, JNK, p38 and horseradish peroxidase (HRP)-conjugated secondary antibody (goat anti-rabbit and goat anti-mouse) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Cell culture Primary ventricular cardiac myocytes were prepared as previously described[27]. Briefly, the hearts from 1–2 d old Sprague-Dawley rats were removed, the ventricles were pooled, and the ventricular cells were dispersed by digestion with collagenase II. The cells were pre-plated for 1 h to enrich the culture with myocytes (90%–95% of cells after this step). Then the cells were cultured in media consisting in high glucose DMEM, 2 mmol/L L-glutamine (GIBCO, Grand Island, NY, USA), penicillin (100 U/mL) and streptomycin (100 μg/mL), 10% FBS at 37 ℃ and 5% CO2 in a humidified atmosphere. The cells were added with silibinin and isoproterenol after 48 h with 5% FBS.
Cell growth assay The cytotoxic effects of isoproterenol on cardiac myocytes were measured using MTT assay as described[28]. The cells were dispensed in 96-well flat-bottomed microtiter plates (Nunc, Roskilde, Denmark) at a density of 5×105 cells/well. After 48 h incubation, they were treated with isoproterenol (10 μmol/L) and/or silibinin at various concentrations for 48 h. The viability was calculated as follows:
Viability (%)=(A490,sample–A490, blank)/(A490, control–A490, blank)×100
Terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) assay The TUNEL assay was used for the detection of DNA strand breaks. The detection was carried out according to the instructions of TACS2 TdT-DAB in situ apoptosis detection kit. Briefly, the cells were rinsed once with PBS and fixed in 3.7% buffered formaldehyde at room temperature for 10 min. The fixed sections were pretreated with 10% H2O2, and end-labeling was performed with TdT labeling reaction mix at 37 ℃ for 1 h. Nuclei exhibiting DNA fragmentation were visualized by incubation in 3,3'-diamino benzidine (DAB) for 7 min. Finally, the sections were counterstained with methyl green, and observed by light microscopy. The nuclei of the apoptotic cells were stained dark brown; TUNEL-positive cardiac myocytes were determined by randomly counting 100 cells.
PKC activity assay PKC activity assay was carried out according to the instructions of the PepTag non-radioactive protein kinase C assay kit (Promega, Madison, WI, USA). Briefly, the cells were washed once with PBS and lysed in lysis buffer, including 20 mmol/L Tris-HCl, 0.5 mmol/L EGTA, 2 mmol/L EDTA, 2 mmol/L phenylmethanesulfonyl fluoride (PMSF), and 10 mg/L leupeptin (pH 7.5). Assays were then performed at 30 ℃ in a total volume of 25 µL containing 5 µL PKC reaction 5× buffer, 5 µL PLSRTLSVAAK peptide, 5 µL PKC activator, 1 µL peptide protection solution, and 9 µL sample. Reactions were initiated by the addition of the 9 µL sample and terminated after 30 min by incubation of the reaction mixture at 95 ℃ for 10 min. After adding 1 µL of 80% glycerol, each sample was separated by 0.8% agarose gel electrophoresis at 100 V for 15 min.
Western blot analysis After incubation for 48 h, both adherent and floating cardiac myocytes were collected. Western blot analysis was carried out as previously described[29] with some modifications. The cells were lysed on ice in lysis buffer [50 mmol/L HEPES (pH 7.4), 1% Triton X-100, 2 mmol/L sodium orthovanadate, 100 mmol/L sodium fluoride, 1 mmol/L EDTA, 1 mmol/L EGTA, 1 mmol/L PMSF], supplemented with proteinase inhibitors (100 µg/mL aprotinin, 10 µg/mL leupeptin, and 100 µg/mL pepstatin) for 1 h. The protein concentration was determined by Folin assay. The lysate was centrifugated at 16 000×g at 4 ℃ for 10 min; equal amounts of total proteins were mixed in 2× loading buffers [50 mmol/L Tris-HCl (pH 6.8), 2% SDS, 10% 2-mercapto-ethanol, 10% glycerol, and 0.002% bromphenol blue], boiled for 5 min, and run on a 12% or 15% SDS-PAGE. Proteins were electrotransferred onto nitrocellulose membranes and detected with antibodies against Ras, Raf-1, Grb2, MAPK and b-actin, followed by the addition of HRP-conjugated secondary antibody and 3,3'-DAB as the HRP substrate.
Statistical analysis All data represented at least 3 independent experiments and were expressed as mean±SD, unless otherwise indicated. One-way ANOVA (SPSS 11.0 software, SPSS, Chicago, IL, USA) was used for statistical analysis, and P<0.05 was considered statistically significant.
Results
Involvement of tyrosine kinase pathway in the protective effect of silibinin Viability and TUNEL assays were carried out to confirm the involvement of the tyrosine kinase pathway in the silibinin-treated cardiac myocytes with isoproterenol exposure. Treatment with 40 µmol/L PTK inhibitor (genistein), 5 µmol/L Ras inhibitor (manumycin A) or 10 nmol/L Raf-1 inhibitor (GW5074) significantly reversed the protective effects of silibinin (Figure 2; Table 1).
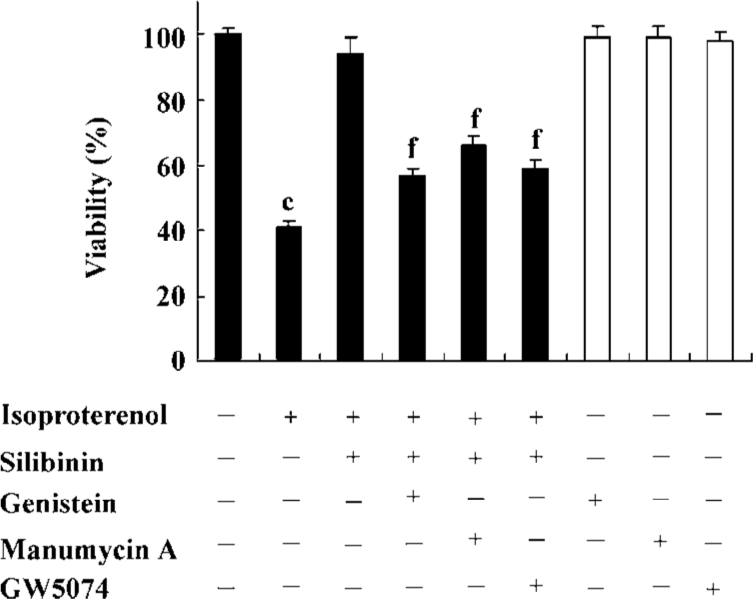
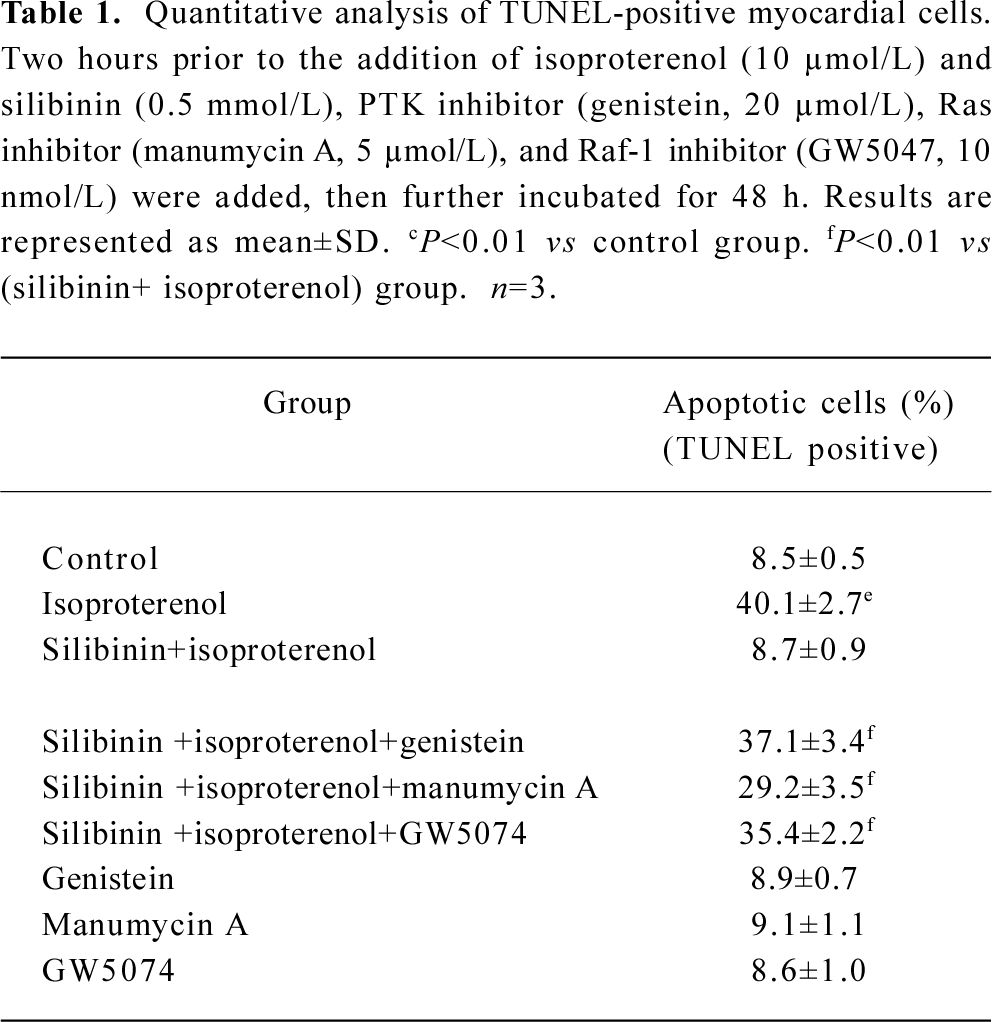
Full table
Ras, Raf-1 and the adaptor protein, Grb2, decreased after incubation with 10 µmol/L isoproterenol for 48 h, whereas silibinin reversed their expression (Figure 4), but after incubation with silibinin alone, the expression of Ras, Raf-1 and Grb2 had no changes, indicating that silibinin could activate the tyrosine kinase pathway when the pathway was inhibited.
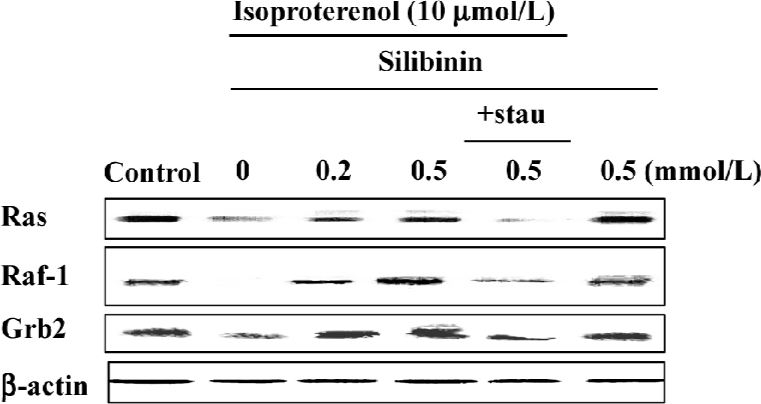
PKC activity increased dose-dependently during silibinin-treatment Very recently, clear evidence has been presented that PKC acts to increase Ras GTP loading, and that the activation of ERK by PKC is Ras dependent[30], thus PKC activity was assayed. The specific fluorescent substrate phosphorylation was observed by treatment with isoproterenol and silibinin. Silibinin dose-dependently increased PKC activity compared to isoproterenol, and PKC activity had no changes after treatment with silibinin alone (Figure 3).
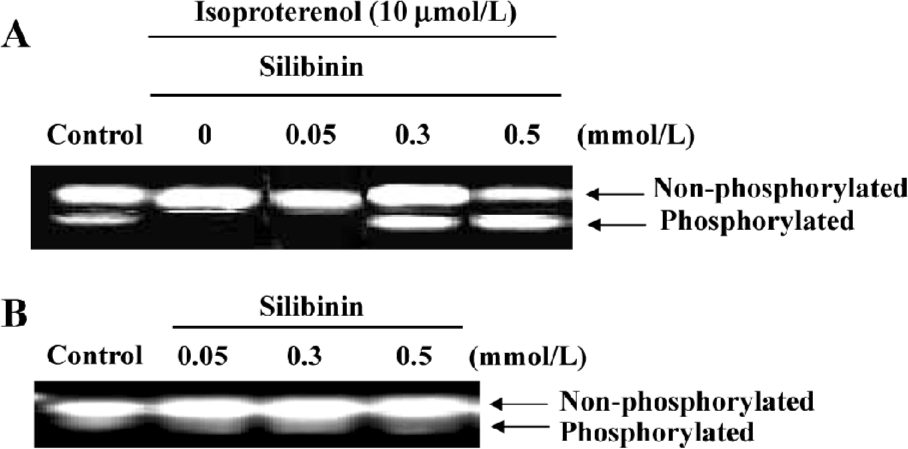
Treatment with 100 nmol/L staurosporine significantly decreased the expression of Ras, Raf-1 and Grb2 (Figure 4). It was suggested that silibinin prevented isoproterenol-induced rat cardiac myocyte apoptosis through the tyrosine kinase pathway after upregulation of PKC activity.
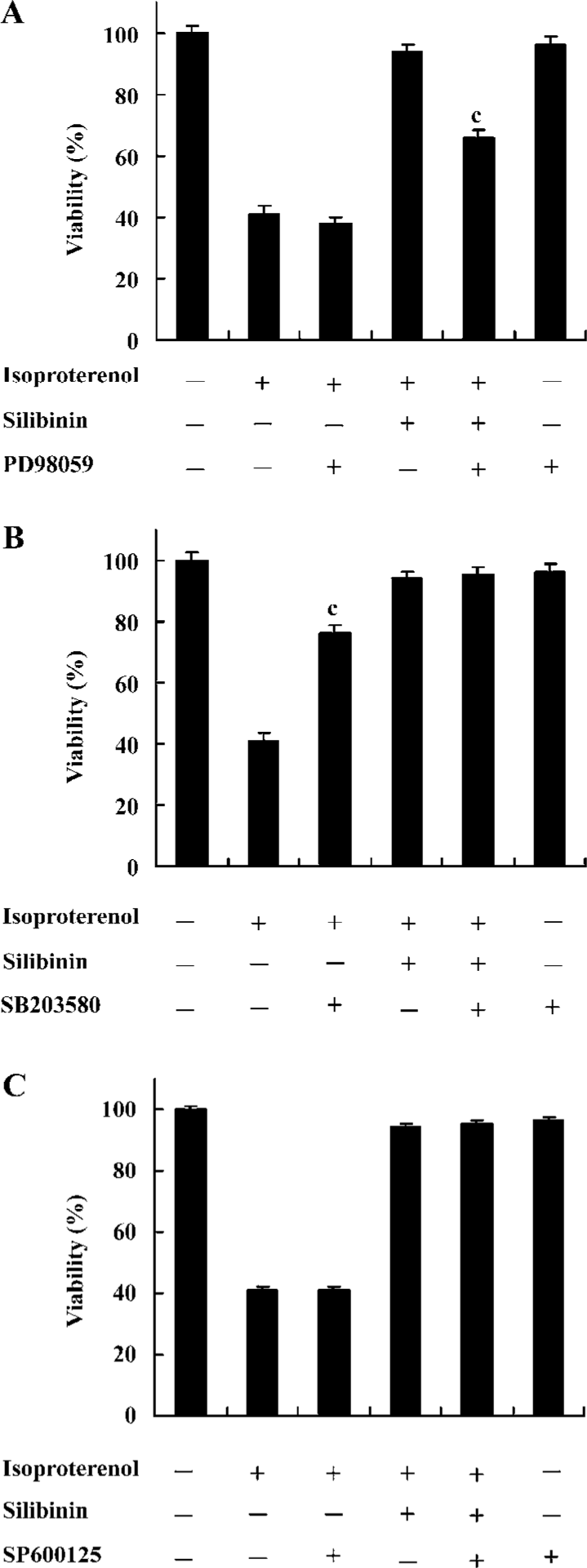
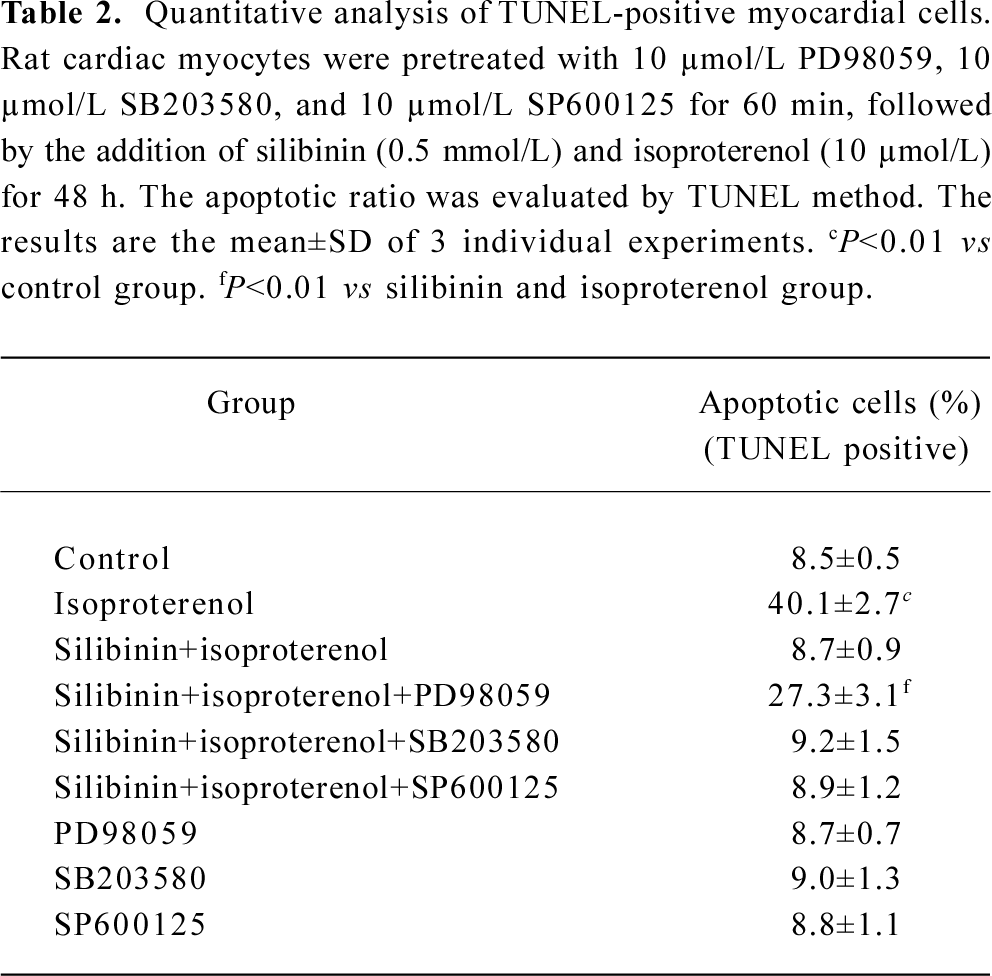
Effects of inhibitors of ERK, p38 and JNK on protective effects of silibinin MAPK cascade is an important signaling modulation pathway that propagates extracellular stimulation into intracellular responses and regulates cell growth, differentiation, and apoptosis[31]. To determine whether MAPK family was involved in silibinin-treatment, 10 μmol/L ERK inhibitor PD98059 (Figure 5A), 10 μmol/L p38 MAPK inhibitor SB203580 (Figure 5B) and 10 μmol/L JNK inhibitor SP600125 (Figure 5C) were administered. Cardiac myocytes were pretreated with 10 μmol/L PD98059, SB203580 and SP600125 for 60 min, and then cultured with silibinin and isoproterenol for 48 h. The protective effect of silibinin was significantly reduced by PD98059, while isoproterenol-induced apoptosis was reversed by SB203580 and both of them were unaffected by SP600125. TUNEL-positive myocardiac cells were increased after inhibitors added, which suggests that the inhibitors could induce myocardiac cells apoptosis (Table 2).
Full table
ERK phosphorylation was increased by silibinin treatment and was dependent on the tyrosine pathway and PKC activity After cardiac myocytes were exposed to silibinin or isoproterenol for 48 h, expression of phosphor-ERK increased (Figure 6A), whereas the expression of phosphorylated p38 decreased compared to isoproterenol alone treatment (Figure 6B), and JNK phosphorylation (Figure 6C) did not change. The expression of ERK and JNK as well as p38 MAPK protein in a whole cell lysate did not change. Ras, Raf-1 and PKC inhibitors caused decrease in ERK phosphorylation, but they had no effect on p38 and JNK phosphorylation (Figure 7). These results suggest that tyrosine kinase pathway and PKC activity affected ERK phosphorylation after treatment with silibinin in rat cardiac myocyte culture. Silibinin had no effect on the activity of MAPKs alone (Figure 6).


Discussion
In response to stress stimulation, cells activate biochemical pathways that allow adaptation to this stressful environ-ment. However, with prolonged isoproterenol treatment, these protective mechanisms may not be sufficient to maintain normal cellular function, and cell injury and death follow. Our previous study showed that silibinin could protect rat cardiac myocytes from apoptosis. In this study, we have demonstrated that genistein, manumycin A and GW5074 significantly reversed the protective effect of silibinin. In addition, the PKC inhibitor, stauroporine, markedly reduced Ras and Raf-1 expression after silibinin treatment. Within the MAPK family members, only ERK was phosphorylated in silibinin treatment and was dependent on the activity of Ras, Raf-1, and PKC.
A central feature of signal transduction downstream of both receptor and oncogenic tyrosine kinases is the Ras-dependent activation of a protein kinase cascade consisting of Raf-1, MEK, and ERK (MAP kinases)[32]. It has been well reported that the family of small G proteins functions as an important link between cell membrane receptors and numerous signaling pathways[33]. Thus, the involvement of small G proteins in cardiac death has become an area of growing interest. Five subfamilies of small G proteins have been described (Rho, Ras, ARF, Rab, Ran), of these, the role of Ras and Rho in cardiac injury have been examined in most detail[34]. Studies of various cell systems have shown that Shc, Grb2, and Sos link activated tyrosine kinases. Potential activators of Raf-1 include protein kinase and activated Ras[35,36]. Our results show that after administration of genistein, manumycin A and GW5074, the protective effects of silibinin were suppressed markedly, indicating that the tyrosine kinase signal cascade including PTK, Ras, and Raf-1 was involved in the enhancement.
The PKC signaling system in cardioprotection has 2 components: (1) the molecules that are involved in PKC signaling; and (2) the manner in which these molecules interact with PKC[37,38]. Evidence from recent studies in numerous cellular theaters suggests that PKC-mediated cardiopro-tection is associated with dynamic modulation of signaling complexes[39]. We further confirmed the PKC activity, which was attenuated by isoproterenol, whereas the impaired activity was reinforced by silibinin. Furthermore, the expression of Ras, Raf-1 and Grb2 were reversed by the PKC inhibitor stauroporine, indicating that PKC was activated in this process, and the activities of Ras and Raf-1 were dependent on PKC.
The Ras and Raf-1 proto-oncogene products are key proteins in the transmission of a variety of intracellular proliferation and differentiation signals. Raf-1 and Ras serve as intermediates in these signaling pathways, connecting upstream tyrosine kinases with downstream serine/threonine kinases such as ERK and MEK[23,24]. This phosphorylation cascade leads to the activation of transcription factors involved in cell growth and differentiation. Raf-1-dependent activation of ERK1 and ERK2 has been demonstrated in wounded intestinal epithelial monolayers[40], mechanically stretched rat cardiac myocytes[41], and shear stress-exposed human endothelial cells[42]. PKC can activate MAPK, which in turn activates the gene transcription involved in cell proliferation and differentiation. Growth factor-mediated activation of the Raf-1–MAPK/ERK cascade may therefore involve either Ras, PKC or both. Our results show that ERK inhibitor, PD98059, in response to silibinin, inhibited the protective effect, whereas the p38 MAPK inhibitor, SB203580, significantly reversed isoproterenol-induced apoptosis; JNK inhibitor, SP600125, had no effect. Western blot analysis further confirmed that phosphorylation of ERK increased with silibinin treatment, but phosphorylation of p38 decreased, and that of JNK did not change, indicating that the protective effect of silibinin required the activation of ERK. Adding Ras, Raf-1 and PKC inhibitors caused a decrease in ERK phosphorylation only, indicating that ERK phosphorylation was dependent on the activity of the tyrosine pathway and PKC.
Silibinin had no effect on the activity of the tyrosine kinase pathway, PKC and MAPK alone, but could activate them when they were inhibited, indicating that silibinin could preserve the balance of kinases in cell homeostasis. Further investigation is necessary to clarify the target of silibinin.
Taken together, the present study shows that silibinin protects isoproterenol-induced apoptosis in rat cardiac myocytes through the activation of PKC involving Ras, Raf-1 and the phosphorylation of ERK.
References
- Berenji K, Drazner MH, Rothermel BA, Hill JA. Does load induced ventricular hypertrophy progress to systolic heart failure? Am J Physiol Heart Circ Physiol 2005;289:H8-H16.
- Frey N, Katus HA, Olson EN, Hill JA. Hypertrophy of the heart: a new therapeutic target? Circulation 2004;109:1580-9.
- Francis GS, Chon JN, Johnson G, Rector TS, Goldman S, Simon A. Plasma norepinephrine, plasma renin activity, and congestive heart failure. Relations to survival and the effects of therapy in V-HeFT II. The V-HeFT VA Cooperative Studies Group. Circulation 1993;87 Suppl 6:VI40-8.
- Communal C, Singh K, Pimentel DR, Colucci WS. Norepinephrine stimulates apoptosis in adult rat ventricular myocytes by activation of the β-adrenergic pathway. Circulation 1998;98:1329-34.
- Li LH, Wu LJ, Zhou B, Wu Z, Tashiro S, Onodera S, et al. Silymarin prevents UV irradiation-induced A375-S2 cell apoptosis. Biol Pharm Bull 2004;27:1031-6.
- Vaziri H, Dessain SK, Eaton EN, Imai SI, Frye RA, Pandita TK, et al. SIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 2001;107:149-59.
- Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, et al. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 2001;107:137-48.
- Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, et al. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science 2004;305:390-2.
- Valenzuela A, Guerra R, Videla LA. Antioxidant properties of the flavonoids silymarin and (+)-cyanidanol-3: comparison with butylated hydroxyanisole and butylated hydroxytoluene. Planta Med 1986;6:438-40.
- Kang JS, Jeon YJ, Park SK, Yang KH, Kim HM. Protection against lipopolysaccharide-induced sepsis and inhibition of interleukin-1β and prostaglandin E2 synthesis by silymarin. Biochem Pharmacol 2004;67:175-81.
- Higmi Y, Shimokawa I. Apoptosis in the aging process. Cell Tissue Res 2000;301:125-32.
- Zhang YP, Herman B. Ageing and apoptosis: mechanisms of ageing and development. Mech Aging Dev 2002;123:245-60.
- Campisi J. Cellular senescence and apoptosis: how cellular responses might influence aging phenotypes. Exp Gerontol 2003;38:5-11.
- Ping P, Zhang J, Zheng YT, Li RC, Dawn B, Tang XL, et al. Demonstration of selective protein kinase C-dependent activation of Src and Lck tyrosine kinases during ischemic preconditioning in conscious rabbits. Circ Res 1999;85:542-50.
- Post GR, Brown JH. G protein-coupled receptors and signaling pathways regulating growth responses. FASEB J 1996;10:741-9.
- Lowy DR, Willumsen BM. Function and regulation of Ras. Annu Rev Biochem 1993;62:851-91.
- Mattingly RR, Macara IG. Phosphorylation-dependent activation of the Ras-GRF/CDC25Mm exchange factor by muscarinic receptors and G-protein βγ subunits. Nature 1996;382:268-70.
- Marais R, Light Y, Paterson HF, Marshall CJ. Ras recruits Raf-1 to the plasma membrane for activation by tyrosine phosphorylation. EMBO J 1995;14:3136-45.
- Jelinek T, Dent P, Sturgill TW, Weber MJ. Ras-induced activation of Raf-1 is dependent on tyrosine phosphorylation. Mol Cell Biol 1996;16:1027-34.
- Fabian JR., Daar IO, Morrison DK. Critical tyrosine residues regulate the enzymatic and biological activity of Raf-1 kinase. Mol Cell Biol 1993;13:7170-9.
- Wang X, Wu BW, Wu DM. Effects of AMP579 and adenosine on L-type Ca2+ current in isolated rat ventricular myocytes. Acta Pharmacol Sin 2005;26:559-62.
- Esposito G, Rapacciuolo A, Naga Prasad SV. Genetic alterations that inhibit in vivo pressure-overload hypertrophy prevent cardiac dysfunction despite increased wall stress. Circulation 2002;105:85-92.
- Kyriakis JM, App H, Zhang XF, Banerjee P, Brautigan DL, Rapp UR, et al. Raf-1 activates MAP kinase-kinase. Nature 1992;358:417-21.
- Tian ZJ, Cui W, Li YJ, Hao YM, Du J, Liu F. Different contributions of STAT3, ERK1/2, and PI3-K signaling to cardiomyocyte hypertrophy by cardiotrophin-1. Acta Pharmacol Sin 2004;25:1157-64.
- Mark L, Hugo AK, Norbert F. Novel molecular targets in the treatment of cardiac hypertrophy. Recent Pat Cardiovas Drug Discov 2006;1:1-20.
- Clerk A, Sugden PH. Untangling the web: specific signaling from PKC isoforms to MAPK cascades. Circ Res 2001;89:847-9.
- Kaburagi S, Hasegawa K, Morimoto T. The role of endothelin converting enzyme-1 in the development of α1-adrenergic-stimulated hypertrophy in cultured neonatal rat cardiac myocytes. Circulation 1999;99:292-8.
- Lasek W, Wankowicz A, Kuc K, Feleszko W, Golab J, Giermasz A. Augmentation of antitumor efficacy by the combination of actinomycin D with tumor necrosis factor-alpha and interferon-gamma on amelanoma model in mice. Cancer Immunol Immuno-ther 1995;40:315-21.
- Suzuki K, Hino M, Kutsuna H, Hato F, Sakamoto C, Takahashi T. Selective activation of p38 mitogen-activated protein kinase cascade in human neutrophils stimulated by IL-1β. J Immunol 2001;167:5940-7.
- Marais R, Light Y, Mason C, Paterson H, Olson MF, Marshall CJ. Requirement of Ras-GTP/Raf complexes for activation of Raf-1 by protein kinase C. Science 1998;280:109-12.
- Clerk A, Pham FH, Fuller SJ. Regulation of mitogen activated protein kinases in cardiac myocytes through the small G protein Rac1. Mol Cell Biol 2001;21:1173-84.
- Wang L., Proud CG. Ras/Erk signaling is essential for activation of protein synthesis by Gq protein-coupled receptor agonists in adult cardiomyocytes. Circ Res 2002;91:821-9.
- Clerk A, Sugden PH. Small guanine nucleotide-binding proteins and myocardial hypertrophy. Circ Res 2000;86:1019-23.
- Sugden PH, Clerk A. Regulation of ERK subgroup of MAP kinase cascades through G protein-coupled receptors. Cell Signal 1997;9:337-51.
- Buday L, Downward J. Epidermal growth factor regulates p21ras through the formation of a complex of receptor, Grb2 adapter protein, and Sos nucleotide exchange factor. Cell 1993;73:611-20.
- Lowenstein EJ, Daly RJ, Batzer AG, Li W, Margolis B, Lammers R, et al. The SH2 and SH3 domain-containing protein Grb2 links receptor tyrosine kinases to ras signaling. Cell 1992;70:431-42.
- Bolli R. The late phase of preconditioning. Circ Res 2000;87:972-83.
- Tong H, Chen W, Steenbergen C, Murphy E. Ischemic preconditioning activates phosphatidylinositol-3-kinase upstream of protein kinase C. Circ Res 2000;87:309-15.
- Ping P, Zhang J, Pierce WM Jr, Bolli R. Functional proteomic analysis of protein kinase C signaling complexes in the normal heart and during cardioprotection. Circ Res 2001;88:59-62.
- Goke M, Kanai M, Lynch-Devaney K, Podolsky DK. Rapid mitogen-activated protein kinase activation by transforming growth factor α in wounded rat intestinal epithelial cells. Gastroenterology 1998;114:697-705.
- Yamazaki T, Komuro I, Kudo S, Zuo Y, Shiojima I, Mizuno T, et al. Mechanical stress activates protein kinase cascade of phosphorylation in neonatal rat cardiac myocytes. J Clin Invest 1995;96:438-46.
- Takahashi M, Berk BC. Mitogen-activated protein kinase (ERK1/2) activation by shear stress and adhesion in endothelial cells. J Clin Invest 1996;99:2623-31.