
Abrupt changes in FKBP12.6 and SERCA2a expression contribute to sudden occurrence of ventricular fibrillation on reperfusion and are prevented by CPU860171
Introduction
Sudden cardiac death (SCD) unexpectedly occurs in patients attributed to cardiomyopathy (CM), heart diseases, or inherited gene mutations[1–3]. It is a serious medical problem as the annual incidence of SCD in the general population is estimated to be as high as 1 in 1000[4]. Etiological factors for SCD varied, including ischemic heart disease, hypertrophied and dilated CM, and genetic defect in ion channels[5]. Patients with congenital mutations of K+ and Na+ channels (abnormality of ion channels in sarcolemma) manifest long QT syndrome (LQTS, mainly LQT1, LQT2, and LQT3) to be at risk for SCD. On the other hand, mutations of the ryanodine type 2 receptor (RyR2, abnormality of the calcium handling system in sarcoplasmic reticulum) exhibit catecholaminergic, polymorphic ventricular tachycardia (CPVT)[6], which is susceptible to develop ventricular fibrillation (VF). Both defective RyR2, attributed to the dissociation of FKBP12.6 by overphosphorylation[7,8] and the disorder of ion channels in the sarcolemma[5], are implicated in mechanisms underlying the development of VF. It is interesting that arrhythmias do not happen in people with disorders of ion channels (channelopathy) at resting stage. But arrhythmias occurred when they were under stimulation of stress or other trigger factors. There are 2 stages of channelopathy in the myo-cardium, as we proposed previously[9], involved in the manifestation of VF, of which a resting state of channelopathy in the myocardium is arrhythmia/VF free, and an activated (deteriorated) state of channelopathy manifests malignant arrhythmias or VF[9]. Thus, we hypothesize that changes in some molecules may abruptly take place within 1−2 min in the diseased myocardium under stress, therefore, VF occurred rapidly. To our knowledge, such changes in molecules responsible for the sudden appearance of VF have not been explored yet.
To observe the occurrence of VF within a limited time, ischemia/reperfusion episode is adopted. An incidence of VF can be observed easily on reperfusion within 10 min. Additionally, we allowed 10 min for coronary occlusion, which is shorter than usual practice, leading to a very low to zero incidence rate of VF on reperfusion in normal rat hearts. During the ischemic period of thyroxin-induced CM, the incidence of VF, is substantially high on reperfusion[10]. The CM caused by L-thyroxin manifests an enhanced ICa.L[11,12] and prolonged action potential duration (APD), which resembles that of inherited LQTS. We also discovered an enhancement of IKr, IKs, and IK1 in isolated myocytes from the CM[13,14]. These alterations are similar in the findings of patients suffering from short QT syndrome (SQTS)[15]. Thus, the CM by thyroxin possesses a strong tendency to develop exaggerated VF, which is probably related to the variability of APD in the myocardium. Such an affected heart is suitable in the investigation of SCD in relation to the abrupt changes in some biological molecules to ventricle fibrillation (VF). It is interesting to investigate a possible link at the molecular level between the rapid occurrence of VF and abrupt changes in the expression of FKBP12.6, SERCA2a, and the endothelin (ET) signaling system in the myocardium[16].
CPU86017, a complex class III antiarrhythmic agent derived from berberine[17], suppresses IK (IKr, IKs) and ICa.L currents in the sarcolemma. Changes in the expression of inflammatory factors, the ET system, and FKBP12.6 in the myocardium were seen in association with the rapid appearance of VF on reperfusion of the CM and suppressed by darusentan[16].
We hypothesized that channelopathies may be divided into 2 stages: the resting stage and the deteriorated stage. Before and during ischemia, the CM possesses ion channelopathy, but no VF is manifested; this is referred to as the resting stage of channelopathy. On reperfusion, the channelopathy is deteriorated, leading to the rapid appearance of VF which may be triggered by abrupt changes in some molecular expressions. We intended to search for molecular changes that are necessary in promoting the deterioration of channelopathy relevant to the manifestation of VF within a very short time on reperfusion. These may include abrupt changes in the expression of FKBP12.6, SERCA2a, PKA, and the ET system in the myocardium. To test the hypothesis, the purpose of the rat CM model was to investigate the transition from ischemia to reperfusion if VF and further molecular changes rapidly appeared on reperfusion. CPU86017, possessing multi-channels blocking and antioxidant activity, was applied to verify the theoretical consideration, and whether it suppresses VF by reversing these changes on reperfusion of the CM.
Materials and methods
Reagents and instruments CPU86017 (Figure 1) is synthesized by the Center of the New Drug Research and Development of the China Pharmaceutical University (Nanjing, China). The RT-PCR reagents were brought from Promega (USA); L-thyroxin was from Sigma (St Louis, MO, USA), the thermal cycler was from Eppendorff (Germany), and Labworks imaging acquisition and analysis software was from Ultra-Violet Products (Cambridge, UK).
L-thyroxin induced CM and reperfusion induced VF The CM was produced as described in previous research[10]. Briefly, male, adult Sprague-Dawley rats (220±20 g) were divided into 5 groups (n=6): the normal control, the CM untreated, CM with coronary artery ligation (CAL), CM+CAL+reperfusion (RF) group, and CM+CAL+RF+ CPU86017 group. The CM group was produced by 0.4 mg/kg sc L-thyroxin for 10 d; CPU86017 was medicated at 4 mg/kg, sc from d 6 to d 10, respectively. On d 11, the rats were anaesthetized with 20% urethane (1.5 g/kg, ip) and CAL was performed for 10 min, followed by reperfusion in rats for 10 min under room temperature at 25 ℃. The lead II of the electrocardiography (ECG) traces was monitored and the absolute irregular waves in ECG were recognized as VF. The hearts were rapidly excised and divided into the right and left ventricle (LV), including the septum, and weighed. The cardiac weight index was measured by heart weight/body weight and LV weight/body weight. The mass of the LV was stored in liquid nitrogen before assay for RT-PCR and Western blotting.
Calcium transients The cardiomyocytes were isolated from the CM and those that were rod-shaped and striated were selected for the calcium transient assay. The assay of [Ca2+]i was performed as described[18,19] with some modifica-tions. After being loaded with 10 µmol/L Fluo-3/AM at 37 ℃ for 30min, unincorporated dye was washed away; the cells, control cardiomyocytes, CM cardiomyocytes, and the CM cardiomyocytes treated with CPU86017 1 µmol, were then transferred to a 300 mL perfusion chamber at room temperature under an IX71 inverted microscope (Olympus, Japan). The myocytes were subjected to 0.5 Hz field stimulation with square wave pulses of 40 V at 5 ms duration. Fluo-3 fluorescence was recorded and converted into absolute [Ca2+]i by a calibration procedure as follows:
[Ca2+]i=Kd*(F - Fmin)/(Fmax - F)
Where Kd represents the dissociation constant for Ca2+-bound to Fluo-3, and F=Fcell - Fback. Fcell and Fback is the measured mean fluorescent intensity of myocytes and background, respectively. Fmax represents the maximum fluorescence of cells permeable to Ca2+ by adding 5 mmol/L MnCl2 to 5 mmol/L A23187-treated myocytes and calculated as follows: Fmax=FMn*5 and Fmin=Fmax/40. The systolic and diastolic [Ca2+]i levels can be calculated from this formula.
RT-PCR analysis The total RNA was extracted from a 100 mg, frozen, left ventricular tissue sample by using 1 mL Trizol and reverse-transcribed into cDNA by AMV reverse transcriptase. The primers in parallel with GAPDH were amplified and assayed semiquantitatively. Oligonucleotides for the primers used are listed in Table 1. The density of the bands was analyzed by computer-aided Labworks imaging acquisition and analysis software. The relative densities of the bands for each mRNA were yielded by dividing the band with the internal control GAPDH.

Full table
Western blotting For the quantitative analysis of total protein levels of FKBP12.6, phospholamban (PLB), and SERCA2a in cardiac myocytes, the LV tissue (100–200 mg) was homogenized in 4 volumes of extraction buffer and centrifuged at 10 000×g for 10 min, and performed as previous described[20]. After transferring to the nitrocellulose and blocking with nonfat milk (5% w/v), followed by incubation with first antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA), sc-6173 for FKBP12s, sc-20511 for PLB and Affinity Bioreagents, MA3-919 for SERCA2a, 1:500 dilution in 5% milk-TBS-Tween) for another hour. After 3 washes, the blot was incubated with horseradish peroxidase-conjugated goat secondary antibody IgG (1:1000) for an additional 1 h. The antigen was detected with 0.1% 3, 3'-diamino-benzidine/0.01% H2O2. A linear relationship between the density of the blots and the protein load was observed when 20, 40, 60, 80, and 100 µg membrane protein were used per lane.
Statistical analysis GraphPad Instat version 3.05 (GraphPad Software, San Diego, CA, USA) was used to analyze the results. Data were presented as mean±SEM. Statistical analyses were performed by ANOVA, followed by a Bonferroni t-test. Fisher’s exact test (one-tailed) was used to test the incidence of VF. A value of P<0.05 was considered statistically significant.
Results
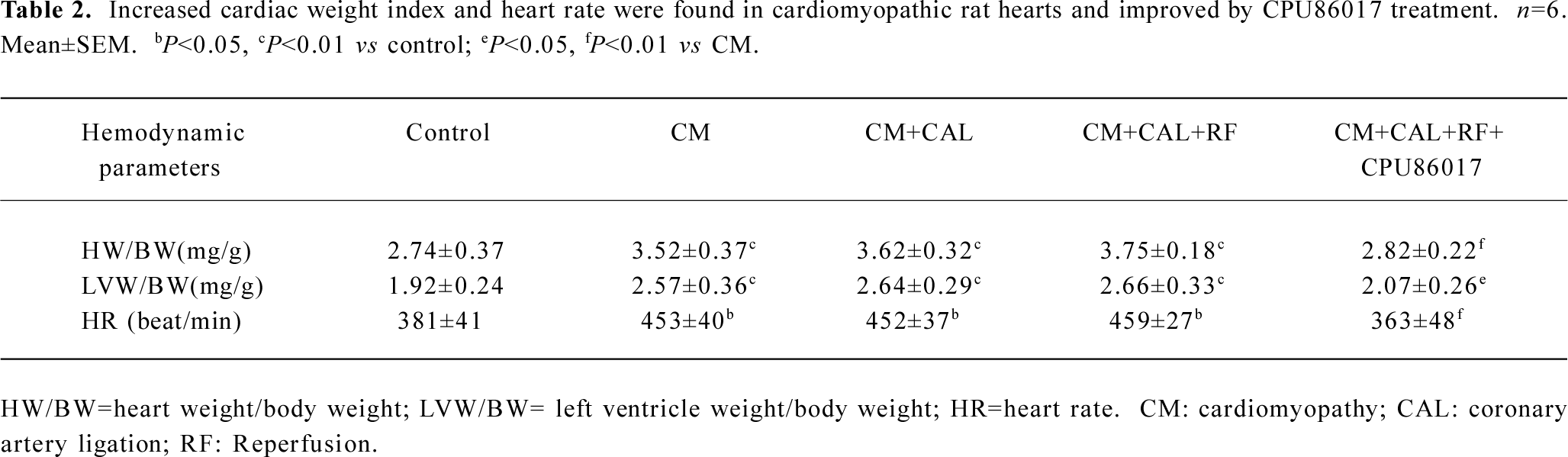
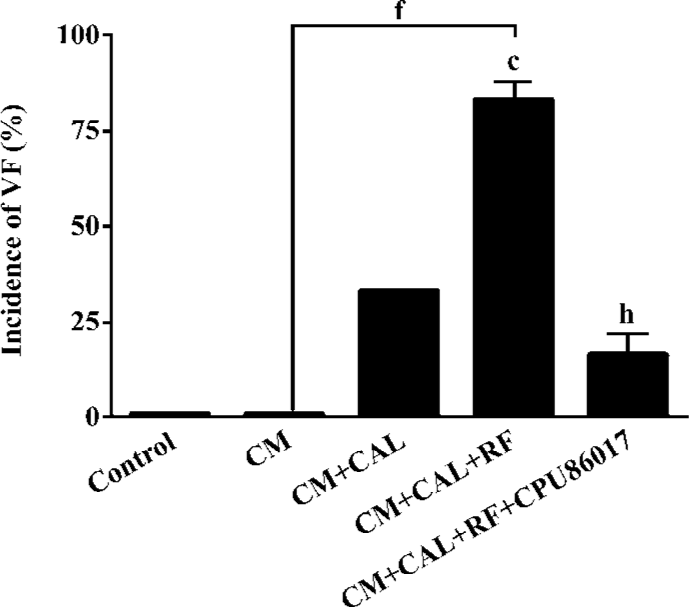
Ventricular hypertrophy and an incidence in VF on reperfusion The cardiac weight index and heart rate (HR) significantly increased in the CM relative to the control. An increased HR was attributed to the stimulation of the beta-adrenergic receptors by chronic thyroxin medication (Table 2). There was no VF observed in the CM before and during CAL. Within 1–2 min following reperfusion, an increased incidence of VF was seen relative to the control and CM groups (P<0.01, Fisher’s exact test, one-tailed). Following intervention with CPU86017, VF was markedly suppressed (P<0.05, Fisher’s exact test, one-tailed) in association with a regression in cardiac hypertrophy (Figure 2).
Full table
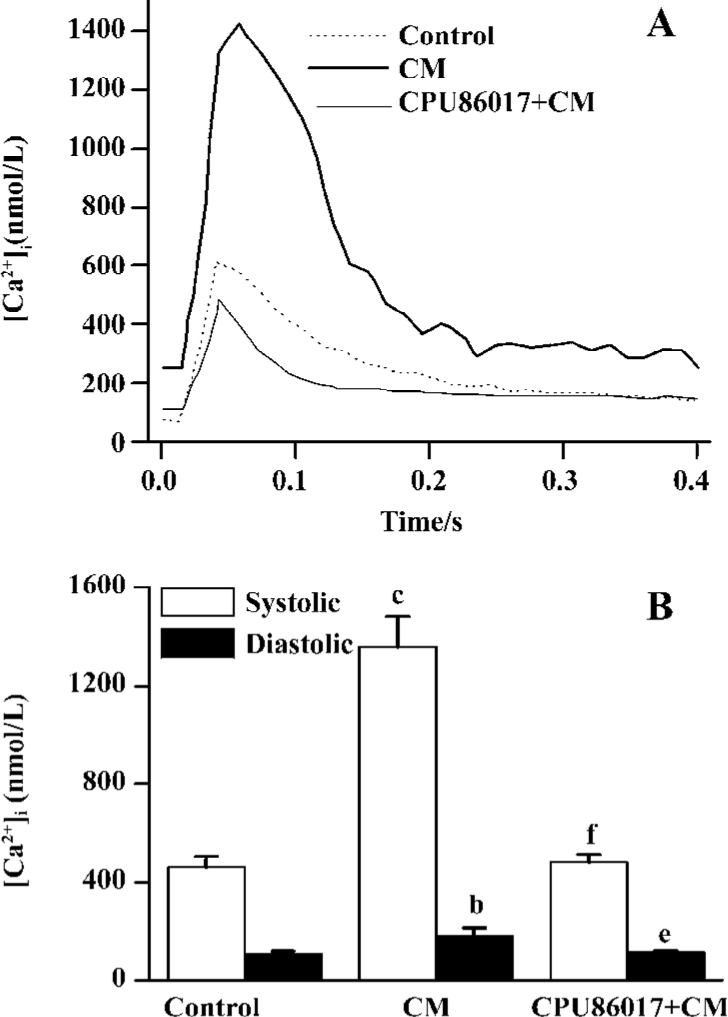
Exaggerated Ca2+ transients and elevated diastolic Ca2+ To test whether an abnormal calcium homeostasis contributes to augmented VF in CM, cardiomyocytes freshly isolated from the CM were tested for calcium transients. A huge peak level (Ca2+ in systole) of intracellular calcium was found in CM (1360±120 nmol/L), relative to the control (460±40 nmol/L, P<0.05, Figure 3A) and provided evidence of dysfunction of the intracellular calcium handling system in the CM. An increase in the trough of [Ca2+]i levels (Ca2+ in diastole, 180±32 nmol/L, Figure 3B) which increases significantly (P<0.05) relative to the control (107±11 nmol/L), indicated that an elevated diastolic calcium served as a risk factor to an increased incidence of VF. Treatment with CPU86017 significantly reduced systolic (480±30 nmol/L) and diastolic (112±5 nmol/L) [Ca2+]i, P<0.05) compared with the untreated CM, respectively.
Abnormal expression of the calcium handling system and PKA For further investigation of the abnormality of the calcium handling system, the mRNA expression of the system was conducted. The upregulation of the RyR2 mRNA level was markedly increased by +82% in the CM and CM+CAL groups (P<0.05, vs control), respectively. These were not further changed following reperfusion (Figure 4A). FKBP12.6 mRNA was significantly was downregulated in the CM and unchanged during 10 min CAL; however, further downregulation was found on reperfusion, (P<0.05 vs CM+CAL+RF, Figure 4B). The mRNA of SERCA2a and PLB was downregulated (P<0.05) in the CM group, relative to the control, respectively, and no further changes were found by ischemia and reperfusion (Figure 4C, 4D). The mRNA expression of PKA was significantly upregulated (P<0.01, vs control) in association with changes in the mRNA of the calcium handling system. Further upregulation of the PKA mRNA expression was found on reperfusion against the untreated CM (P<0.05, Figure 4E). This abnormal expression of the calcium handling system and PKA were completely reversed by CPU86017 treatment (P<0.01).
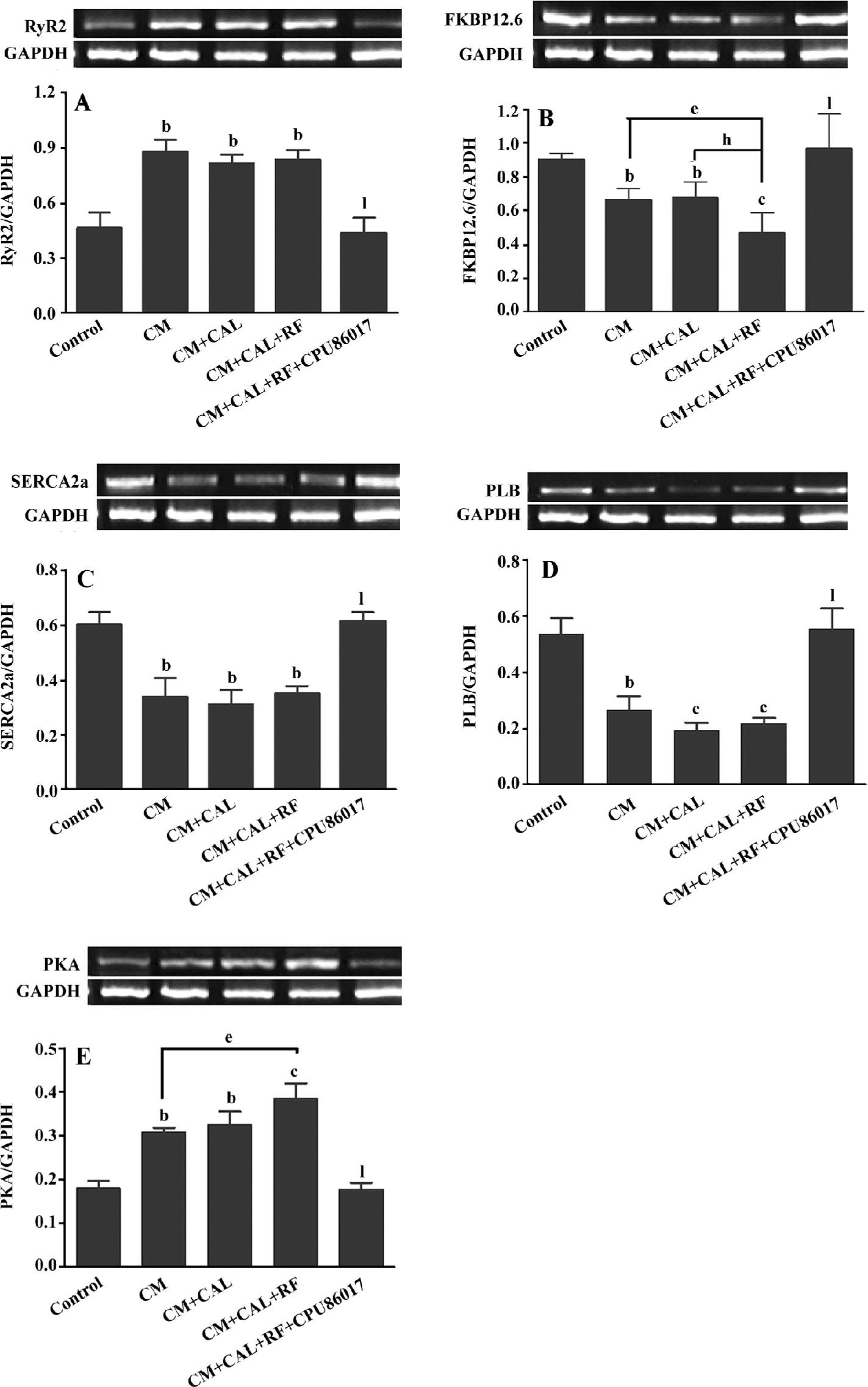
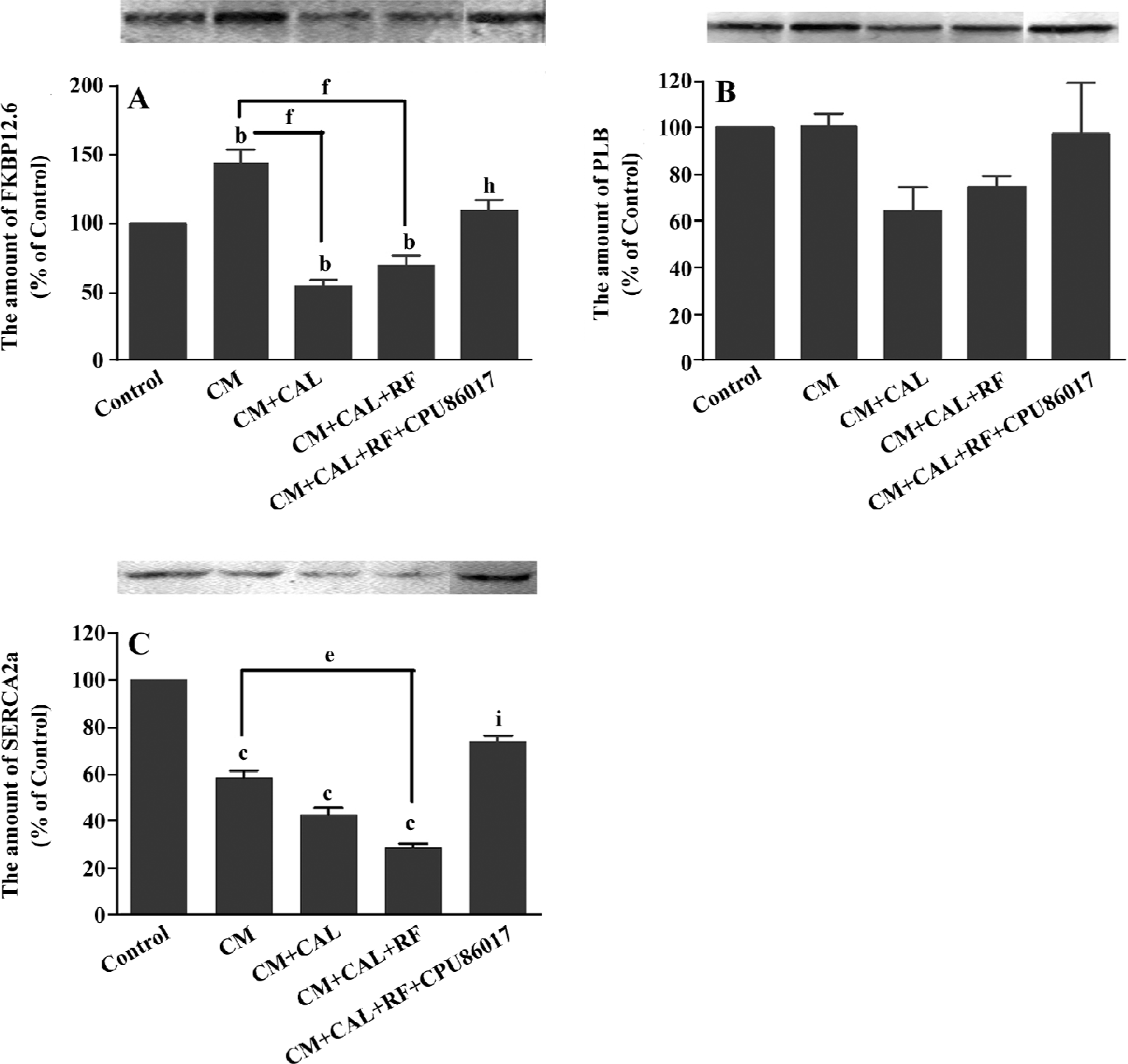
During CAL and RF, the protein expression of FKBP12.6 was downregulated significantly relative to the CM (P<0.05); however, the expression of FKBP12.6 before ischemia in the CM was upregulated relative to the control (P<0.01, Figure 5A). The protein levels of SERCA2a significantly decreased in the CM group (P<0.01 vs control) and further downregulation was found on reperfusion (P<0.01 vs control, Figure 5B). The change in the PLB protein level was mild and no difference was found among the groups (Figure 5C). CPU86017 significantly regressed the downregulation of the protein expression of FKBP12.6 and SERCA2a, respectively (P<0.01 vs CM+CAL+RF).
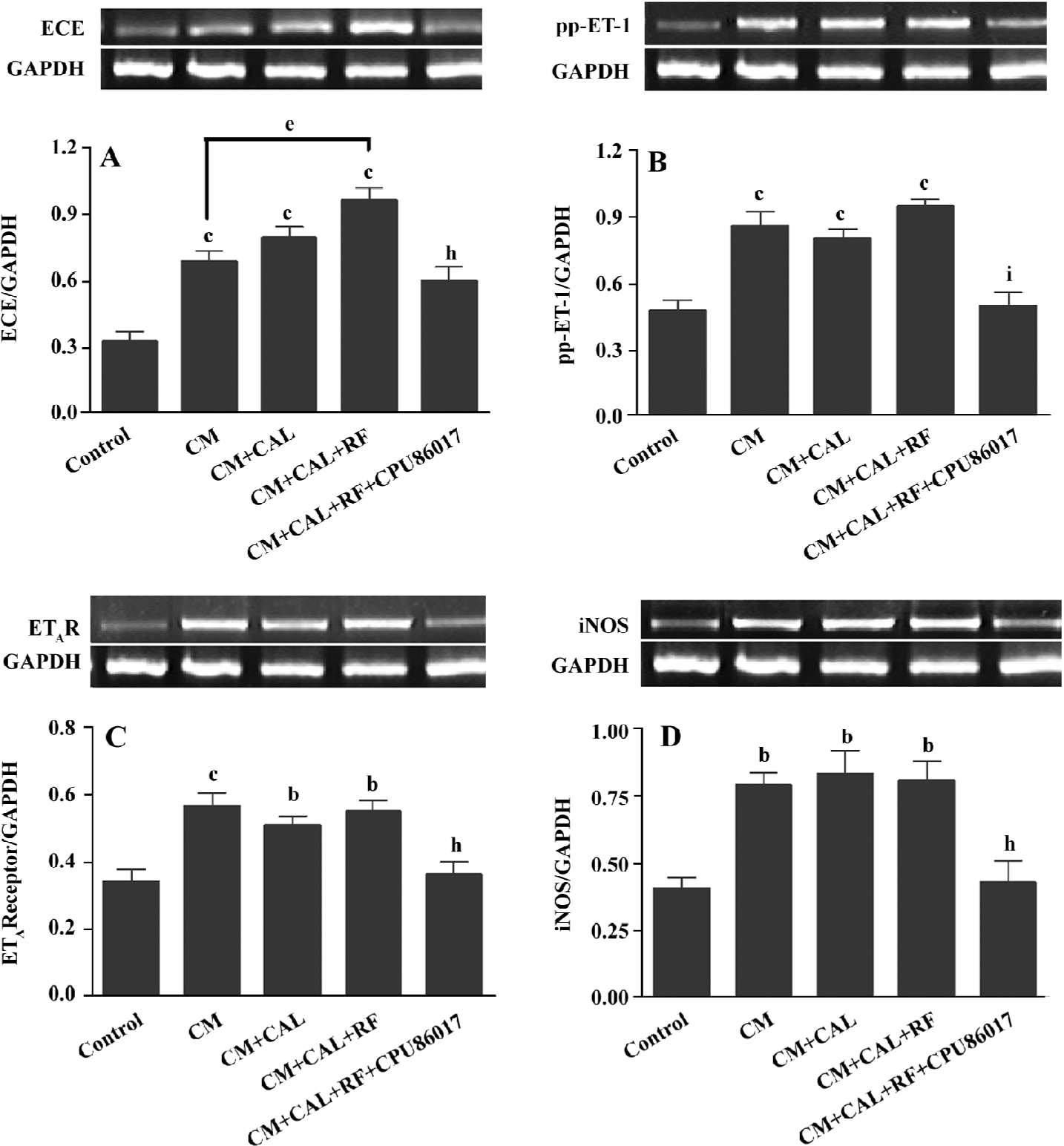
Overactivated ET system and upregulated iNOS The ET-converting enzyme (ECE), prepro-ET-1 (pp-ET-1), endothelin A receptor (ETAR), and iNOS mRNA expression was significantly upregulated in the CM, CM+CAL and CM+CAL+RF groups (P<0.05 vs control, Figure 6), but no difference was found between ischemia and reperfusion, except that the mRNA of ECE was further upregulated on reperfusion (P<0.05, Figure 6A). CPU86017 completely reversed these abnormalities in the CM.
Discussion
Sudden augmented VF has been found on reperfusion (CM+CAL+RF) relative to the ischemia (CAL+CM) group. The expression of some molecules changed rapidly on reperfusion of the CM, relative to the CM or CAL; among them, the protein expression of FKBP12.6 and SERCA2a downregulated significantly relative to the CM (P<0.05). In contrast, no change in RyR2 and PLB was found. These changes in the calcium handling system are associated with an increased rate of VF on reperfusion. A suppression on abnormal expression by CPU86017 results in the disappearance of VF during the 10 min of reperfusion.
Characteristics of dysfunction of the RyR2 in cardio-myocytes freshly isolated from the CM reveals a great increment in systolic [Ca2+]i. A significant peak of calcium is attributed to an increased release by RyR2, and is partly attributed to an increase in Ca2+ influx via ICa.L by L-thyroxin treatment[11,12]. The abnormality of RyR2 is likely to develop a calcium leak which elevates diastolic [Ca2+]i and delays the repolarization in the myocardium so that it is favorable to arrhythmogenesis[21]. Besides the unstable RyR2[8], an insufficient calcium pump attributable to the downregulation of SERCA2a may be implicated[7]. On reperfusion, a dramatic further downregulation of FKBP12.6 and SERCA2a deteriorates the calcium homeostasis in the diastole to retard the repolarization process, thus, a non-reentrant mechanism is triggered to help a process of the delayed after depolarization (DAD), torsades de pointes, and finally VF[8,22].
An upregulation of RyR2 mRNA in this study is in agreement with findings of previous reports[16,23]. However, an upregulation of mRNA of RyR2 in the CM is at odds with those of the post-myocardial infarcted rat heart stimulated by isoproterenol[24], in a failing heart[8], diabetic CM[20,25], and CM by chronic anthracycline administration[26] where the downregulation of RyR2 mRNA is remarkable. The upregulation of RyR2 and FKBP12.6 in the CM before reperfusion may be explained by increased transcription in the nucleus by L-thyroxin. Another possible reason of an upregulation of FKBP12.6 in the CM may be related to the use of the polyclonal antibody which conjugates the total protein of FKBP12, including the subunit FKBP12.6[37].
An activated endothelin signaling pathway is implicated in cardiovascular dysfunction and disorders[28] and presumably exerts a role in deteriorating channelopathy of the SERCA2a and RyR2. The upregulation of the mRNA of pp-ET-1, ECE, and ETAR is found in the CM before and during ischemia, manifesting no VF; however, an abrupt upregulation of ECE mRNA is associated with VF on reperfusion indicating that an activated ET signaling system may play an active part in the rapid manifestation of VF, coinciding with findings in a previous report[16]. The upregulation of the iNOS gene expression is related to the genesis of the reactive oxygen system (ROS) which combines an activated ET pathway to form an ET-ROS signaling pathway[29]. The ROS injures the Ca2+ handling function and gene expression on reperfusion[30] and promotes the formation of an activated NFκB and TNFα, both of which facilitate the transcript process in the nucleus responsible for an upregulation of the ET system, and then an increase in the susceptibility of the CM to arrhythmogenesis. An excess of ROS is suppressed when an activated ET pathway is antagonized[31]. Oxidative stress is augmented by thyroxin treatment and suppressed by CPU86017[32], thus, antioxidative activity of CPU86017 does supplement a relief of the deteriorated channelopathy on reperfusion.
Increased calcium in the diastole mediates triggered activity to initiate DAD, and eventually malignant tachyventricular arrhythmias[24]. Compound JTV-519, a derivative of diltiazem, possesses a blockade on calcium and potassium channels[33] and has been discovered to correct deficiencies of RyR2 and FKBP12.6 effectively[34]. Its profile resembles those of CPU86017. A calcium antagonism has been found clinically to improve the lifespan of patients who suffer from CPVT[35].
APD, or the process of repolarization, depends on the balance of total inward and outward ion currents and an increment in IKr or IKs which has been discovered in the CM, facilitating the repolarization process to shorten APD[13,14]. SQTS from mutated genes exhibits an increase in IKr, IKs, or IK1 by “gain of function” mutation, which predisposes patients to life-threatening arrhythmias[36]. Thus, the CM may possess a reentry mechanism resembling SQTS. Additionally, an enhanced ICa.L current in the CM[11,12] produces a prolonged APD indicating that a non-reentrant mechanism is likely to be involved. Thus, the variability of repolarization in the CM serves as a basis for predisposition to severe cardiac arrhythmias in stress[37,38]. An abnormality of the expression of the calcium handling system may be associated with the variability of repolarization, eventually contributing to the appearance of VF in stress.
An exacerbated VF incidence is rapidly found on reperfusion against a state of VF free of the CM and during 10 min ischemia. Reperfusion acts as stress to produce molecular changes for the exacerbation and appearance of VF. Theoretically, 2 stages of channelopathy[9] or multi-hit concept[3,39] have been proposed to explain the complicated processes underlying mechanisms in SCD. A patient with mutated genes does not exhibit cardiac arrhythmias, but is potentially at risk to develop severe cardiac arrhythmias[40]. The frequency of genetic mutations of individual channels as SCN5A or CPVT is much higher, up to 10%–20% of the population, than those to manifest LQTS or the CPVT. This phenomenon supports the concept that the manifestation of LQTS and CPVT needs molecular activation of ion channelopathy. It is necessary to make such a conversion in CM from the resting state to the deteriorated stage. The deterioration of channelopathies in molecular aspects contributing to the SCD or malignant arrhythmias, or the manifestation of LQTS, can be triggered by either physical and mental stress or drug insults[25,40,41].
SCD is always abrupt, unpredictable, and unpreventable; however, an awareness of molecular activation at the upstream to channelopathy responsible for the sudden appearance of malignant arrhythmias hopefully provides a way to prevent patients from the SCD. Thus, the upstream in the signaling pathways and transcription process in the nucleus has been the center of focus, rather than the limitation to the ion channels, and these targets are helpful in searching for effective agents to prevent the SCD. Based on the findings of this study, we suggested that reperfusion produces oxidative stress which causes biosynthesis and the release of more ET, therefore, the facilitation of PKA upregulation and abnormality of the calcium handling system and the ion channels in sarcolemma is the result. An augmented ICa.L channel is attributed to PKA (presumably including PKC) phosphorylation relating to an activated ET system[42] and arrhythmogenesis[24].
In the CM there may be a link (which may be an upregulation of PKA or PKC) between augmented IKr, IKs, and ICa.L currents and abnormal expression of FKBP12.6, RyR2, SERCA2a, and PLB. PKA and PKC are both responsible for the predisposition of the hearts to malignant arrhythmias. Reperfusion facilitates molecular events which include an activation of NF-κB, mitogen activated protein kinase (MAPK), and AP-1[16,43] to increase the transcription process for the upregulation of these genes encoding ECE and PKA. Reperfusion also causes variability of the repolarization process in the myocardium, thus, a blockade by CPU86017 on IKr and IKs suppresses the shortening of APD in the CM (data not shown); the ICa.L blocking effect of CPU86017 inhibits a prolongation of APD in the myocardium[17]. Eventually, as a result, the susceptibility of the CM to VF is dramatically relieved by CPU86017.
In conclusion, abrupt molecular changes contributing to the sudden occurrence of VF within 1–2 min of reperfusion has been discussed. A swift downregulation of FKBP12.6 and SERCA2a, and the upregulation of PKA and the ET signaling pathway, are the main events relevant to manifest VF. We also demonstrated a conversion of a silent resting state into an activated channelopathy contributing to the occurrence of VF. The CM by thyroxin is applied to induce basic channelopathy (the resting state), but VF free and the sudden manifestation of VF on reperfusion is triggered by dramatic worsening in channelopathies. With a blockade on multi-channels and an anti-oxidative effect, abrupt molecular changes on reperfusion are markedly suppressed by CPU86017, resulting in the prevention of sudden appearance of VF.
References
- Roberts R, Brugada R. Genetics and arrhythmias. Annu Rev Med 2003;54:257-67.
- Dai DZ, Yu F. Ion channelopathy and hyperphosphorylation contributing to cardiac arrhythmias. Acta Pharmacol Sin 2005;26:918-25.
- Rubart M, Zipes DP. Mechanisms of sudden cardiac death. J Clin Invest 2005;115:2305-15.
- Sen-Chowdhry S, McKenna WJ. Sudden cardiac death in the young: a strategy for prevention by targeted evaluation. Cardio-logy 2006;105:196-206.
- Schwartz PJ. Management of long QT syndrome. Nat Clin Pract Cardiovasc Med 2005;2:346-51.
- Tester DJ, Spoon DB, Valdivia HH, Makielski JC, Ackerman MJ. Targeted mutational analysis of the RyR2-encoded cardiac ryanodine receptor in sudden unexplained death: a molecular autopsy of 49 medical examiner/coroner’s cases. Mayo Clin Proc 2004;79:1380-4.
- Olson EN. A decade of discoveries in cardiac biology. Nat Med 2004;10:467-74.
- Wehrens XH, Lehnart SE, Huang F, Vest JA, Reiken SR, Mohler PJ, et al. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell 2003;113:829-40.
- Dai DZ. Two patterns of ion channelopathy in the myocardium: perspectives for development of anti-arrhythmic agents. Curr Opin Investig Drugs 2005;6:289-97.
- Yu F, Dai DZ, An LF, Guo XF. Heart hypertrophy induced by levothyroxine aggravates ischemic lesions and reperfusion arrhythmias in rats. Acta Pharmacol Sin 1997;18:71-4.
- Dai DZ, Hu HJ, Yang DM, Hao XM, Zhang GQ, Zhou PA, et al. Chronic levothyroxin treatment is associated with ion channel abnormalities in cardiac and neuronal cells. Clin Exp Pharmacol Physiol 1999;26:819-21.
- Wang HL, Li SB, Dai DZ. Change on L-type calcium current in single guinea pig hypertrophic ventricular myocytes induced by levothyroxine. J China Pharm Univ 2000;31:130-4.
- Zhang GQ, Hao XM, Ma YP, Zhou PA, Wu CH, Dai DZ. Characteristics of the delayed rectifier K+ current in guinea pig hypertrophied ventricular myocytes induced by thyroxin. China Pharmacol Bull 2001;17:174-7.
- Zhang GQ, Ma YP, Hao XM, Zhou PA, Wu CH, Dai DZ. Rapidly activating delayed rectifier K+ current and inward rectifier K+ current in cardiomyocytes from hypertrophied guinea pig hearts induced by thyroxine. J China Pharm Univ 2000;31:451-4.
- Brugada R, Hong K, Cordeiro JM, Dumaine R. Short QT syndrome. CMAJ 2005;173:1349-54.
- Xia HJ, Dai DZ, Dai Y. Up-regulated inflammatory factors endothelin, NFκB, TNF-α and iNOS involved in exaggerated cardiac arrhythmias in L-thyroxin induced cardiomyopathy are suppressed by darusentan in rats. Life Sci 2006;79:1812-9.
- Dai DZ. CPU86017: a novel class III antiarrhythmic agent with multiple actions at ion channels. Cardiovasc Drug Rev 2006;24:100-14.
- Kao JP, Harootunian AT, Tsien RY. Photochemically generated cytosolic calcium pulses and their detection by Fluo-3. J Biol Chem 1989;264:8179-84.
- Virag L, Scott GS, Antal-Szalmas P, O’Connor M, Ohshima H, Szabo C. Requirement of intracellular calcium mobilization for peroxynitrite-induced poly(ADP-ribose) synthetase activation and cytotoxicity. Mol Pharmacol 1999;56:824-33.
- Qi MY, Xia HJ, Dai DZ, Dai Y. A novel endothelin receptor antagonist CPU0213 improves diabetic cardiac insufficiency attributed to up-regulation of the expression of FKBP12.6, SERCA2a and PLB in rats. J Cardiovasc Pharmacol 2006;47:729-35.
- Katra RP, Laurita KR. Cellular mechanism of calcium-mediated triggered activity in the heart. Circ Res 2005;96:535-42.
- Wehrens XH, Marks AR. Sudden unexplained death caused by cardiac ryanodine receptor (RyR2) mutations. Mayo Clin Proc 2004;79:1367-71.
- Wu XD, Dai DZ, Zhang QP, Gao F. Propranolol and verapamil inhibit mRNA expression of RyR2 and SERCA in L-thyroxin-induced rat ventricular hypertrophy. Acta Pharmacol Sin 2004;25:347-51.
- Wang YQ, Shi YP, Dai DZ. Therapeutic effects of CPU 86017 on acute and chronic congestive cardiac failure mediated by reducing ET-1, NOS and oxidative stress in rats. Drug Dev Res 2004;63:22-32.
- Ligeti L, Szenczi O, Prestia CM, Szabo C, Horvath K, Marcsek ZL, et al. Altered calcium handling is an early sign of strepto-zotocin-induced diabetic cardiomyopathy. Int J Mol Med 2006;17:1035-43.
- Gambliel HA, Burke BE, Cusack BJ, Walsh GM, Zhang YL, Mushlin PS, et al. Doxorubicin and C-13 deoxydoxorubicin effects on ryanodine receptor gene expression. Biochem Biophys Res Commun 2002;291:433-8.
- Jeyakumar LH, Ballester L, Cheng DS, McIntyre JO, Chang P, Olivey HE, et al. FKBP binding characteristics of cardiac microsomes from diverse vertebrates. Biochem Biophys Res Commun 2001;281:979-86.
- Miyauchi T, Masaki T. Pathophysiology of endothelin in the cardiovascular system. Annu Rev Physiol 1999;61:391-415.
- Xu FP, Chen MS, Wang YZ, Yi Q, Lin SB, Chen AF, et al. Leptin induces hypertrophy via endothelin-1-reactive oxygen species pathway in cultured neonatal rat cardiomyocytes. Circulation 2004;110:1269-75.
- Temsah RM, Netticadan T, Chapman D, Takeda S, Mochizuki S, Dhalla NS. Alterations in sarcoplasmic reticulum function and gene expression in ischemic-reperfused rat heart. Am J Physiol 1999;277:H584-94.
- Gupta SK, Saxena A, Singh U, Arya DS. Bosentan, the mixed ETA-ETB endothelin receptor antagonist, attenuated oxidative stress after experimental myocardial ischemia and reperfusion. Mol Cell Biochem 2005;275:67-74.
- Hao JM, Dai DZ, Yu F. The oxidative stress enhancing effects by L-thyroxin and the antioxidant effects of CPU 86017, a derivative of tetrahydroberberine. Prog Pharmaceut Sci 2005;29:417-21.
- Nakaya H, Furusawa Y, Ogura T, Tamagawa M, Uemura H. Inhibitory effects of JTV-519, a novel cardioprotective drug, on potassium currents and experimental atrial fibrillation in guinea-pig hearts. Br J Pharmacol 2000;131:1363-72.
- Kohno M, Yano M, Kobayashi S, Doi M, Oda T, Tokuhisa T, et al. A new cardioprotective agent, JTV519, improves defective channel gating of ryanodine receptor in heart failure. Am J Physiol Heart Circ Physiol 2003;284:H1035-42.
- Sumitomo N, Harada K, Nagashima M, Yasuda T, Nakamura Y, Aragaki Y, et al. Catecholaminergic polymorphic ventricular tachycardia: electrocardiographic characteristics and optimal therapeutic strategies to prevent sudden death. Heart 2003;89:66-70.
- Schimpf R, Wolpert C, Gaita F, Giustetto C, Borggrefe M. Short QT syndrome. Cardiovasc Res 2005;67:357-66.
- Piccirillo G, Magri D, Matera S, Magnanti M, Torrini A, Pasquazzi E, et al. QT variability strongly predicts sudden cardiac death in asymptomatic subjects with mild or moderate left ventricular systolic dysfunction: a prospective study. Eur Heart J 2006. [Epub ahead of print].
- Thomsen MB, Verduyn SC, Stengl M, Beekman JD, de Pater G, van Opstal J, et al. Increased short-term variability of repolarization predicts d-sotalol-induced torsades de pointes in dogs. Circulation 2004;110:2453-9.
- Tomaselli GF, Zipes DP. What causes sudden death in heart failure? Circ Res 2004;95:754-63.
- Gollob MH. Genetic profiling as a marker for risk of sudden cardiac death. Curr Opin Cardiol 2006;21:42-6.
- Chow BJ, Gollob M, Birnie D. Brugada syndrome precipitated by a tricyclic antidepressant. Heart 2005;91:651.
- Woo SH, Lee CO. Effects of endothelin-1 on Ca2+ signaling in guinea-pig ventricular myocytes: role of protein kinase C. J Mol Cell Cardiol 1999;31:631-43.
- Chen J, Mehta JL. Angiotensin II-mediated oxidative stress and procollagen-1 expression in cardiac fibroblasts: blockade by pravastatin and pioglitazone. Am J Physiol Heart Circ Physiol 2006;291:H1738-45.