
Short hairpin RNA-mediated inhibition of HSV-1 gene expression and function during HSV-1 infection in Vero cells
Introduction
RNA-mediated gene suppression was first described for plants as a defense mechanism against viruses and tran-sposons[1]. Subsequently, this mechanism was also described in organisms ranging from yeast to mammals. RNA interference (RNAi) has now opened new avenues in the fields of virology, cancer research, and drug design[2,3]. The mechanism of RNA-inducible gene silencing is activated by a double-stranded RNA molecule (21–25 bp in length) corresponding to the sequence of the target gene to be silenced[4]. For therapeutic application against viral diseases, the small interfering RNA must be able to enter infected cells efficiently and inhibit an ongoing virus infection[5]. In mammals, RNA-mediated gene silencing has not yet been demonstrated to play an active endogenous role in antiviral defense. However, the components of this ancient system have been retained and have contributed to multiple aspects of genome regulation.
Herpes simplex virus type 1 (HSV-1) is a double-stranded DNA virus that infects epithelial and neuronal cells. The envelope of HSV-1 contains at least 6 virus-encoded glycoproteins which are exposed on the virion surface. Glycoprotein D (gD) from HSV-1 is coded by HSV gene US6. It plays a role in membrane-related events involved in virus spread. gD is the entry, receptor-binding protein of the virus[6]. The purpose of this study was to determine whether a short hairpin RNA (shRNA) could suppress the expression of the US6 gene during infection[7]. In the study, we screened 3 shRNA and identified the most efficient one for the HSV-1 mRNA target S gene.
Materials and methods
Cells and viruses The African green monkey kidney cell line (Vero, strain 186) was obtained from Wuhan University Cell Storage Center (Wuhan, China). Cells were grown in Dulbecco’s modified Eagle’s medium (DMEM; Sigma, St Louis, MO, USA) supplemented with 10% neonatal calf serum and 100 μg/mL penicillin and streptomycin in a humidified 5% CO2 incubator at 37 °C. The test medium used for experiments contained only 2% serum. Working stocks of HSV-1 (strain KOS) were propagated at 0.01 plaque-forming units per cell using Vero cells as the host, and prepared using standard procedures.
Generation of shRNA Three shRNA sequences were screened in order to identify the best sequence for silencing an HSV-1 gene target. The KOS strain gD coding sequence targeted by shRNA was identical to the published GenBank strain 17 HSV-1 sequence (NC 001806) and was found to be HSV specific by GenBank BLAST analysis. Targeting sequences were compared against the GenBank database to exclude sequences that may affect cellular genes. The target gene is shown in Table 1. shRNA was transcribed using the MessageMuter™ shRNA production kit (Epicentre, Madison, Wisconsin, USA) according to the manufacturer’s recommendations. Oligonucleotides that contained the T7 RNA polymerase promoter were annealed to a 60 mer oligonucleotide, which contained an antisense T7 RNA polymerase promoter sequence, 21 nucleotides corresponding to the target sequence (Table 1), an 8 nucleotide loop region, 21 nucleotides complementary to the target sequence, and 2 adenine residues (5’AA [sense 21] AACCAGAAGA [antisense 21] TATAGTGA). The ends of the annealed duplex were filled in using the Exo-Minus Klenow DNA polymerase (Epicentre, Madison, Wisconsin, USA) and dNTP to generate a linear double-stranded DNA that served as a T7 in vitro transcription template. shRNA was transcribed from the DNA template in a rapid, high-yield in vitro transcription reaction. The transcribed RNA spontaneously formed a hairpin structure (shRNA) in the solution [8]. Following clean up, shRNA was transfected into cultured cells.
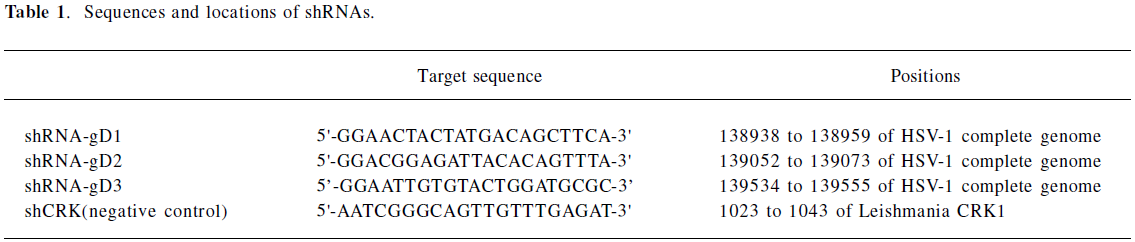
Full table
Plaque assays The infectious titres of HSV-1 were determined by plaque assays. The Vero cells were grown in a 24-well tray to 80%–90% confluence and then transfected with specific or control shRNA (30–180 nmol/L) by Lipofectin complex (RNAiFect transfection reagent; Qiagen, Valencia, CA, USA). After 24 h, the cells were infected with HSV-1 at a multiplicity of infection (MOI) of 10[9]. Cells were overlaid with 2 mL of a 1:1 mixture of l% methylcellulose and 2×DMEM to allow only a cell-to-cell spread of virus. The cells were fixed and stained overnight with a solution of 0.4% crystal violet in a mixture of formalin (3%, v/v) and ethanol (1.67%, v/v) in water, and the numbers of plaques were counted at 2 d postinfection. Overlapping plaques and plaques at the edge of the well were counted as a single plaque for the purpose of deriving the number of plaques per well[9].
Western blotting analysis The expression of gD was detected by Western blotting at 24 and 48 h postinfection[10]. Briefly, the cells were washed twice with ice-cold phosphate-buffered saline (PBS) and lysed in 200 μL of lysis buffer per well in a 6-well plate. The procedure was carried out on ice for 15 min and the supernatants were collected by centrifugation at 12 000×g for 5 min at 4 ºC. The protein concentration was measured by ultraviolet spectrophotometer (Biochrom, Cambridge, UK). Subsequently, 10 μg of total protein from each sample was loaded on 10% SDS–PAGE. For the Western blotting analysis, gels were electroblotted to a nitrocellulose membrane, which was soaked for 1 h and incubated for 1 h at room temperature in the presence of a goat anti-gD polyclonal antibody (Chemicon, CA, USA; 1:1000 dilution). The β-Actin gene (mouse anti-β-Actin monoclonal antibody; Santa Cruz Biotechnology, Santa Cruz, CA, USA; 1:5000 dilution) was used as the internal control. After incubation, the membrane was washed 3 times, and rabbit antigoat conjugated with horseradish peroxidase immunoglobulin G (IgG; Chemicon, CA, USA; 1:10000 dilution) was added, followed by incubation for an additional 1 h. Antibody–antigen complexes were visualized using the EzWay DAB Western blot kit (Koma Biotechnology, Gangseo-gu, Seoul, KORE).
Flow cytometry assay The viral antigen expression in HSV-1-infected cells was analyzed by flow cytometry at 24 and 48 h post infection. The anti-HSV-1 gD monoclonal antibody and goat antimouse IgG–fluorescein isothiocyannate (FITC) (Sigma–Aldrich, St Louis, MO, USA) were used in the assay[11]. Vero cells were grown in 6-well plates and treated as described earlier for the in vitro plaque assays. At the end of experiment, the overlay was removed and the cells were washed with PBS. They were then dissociated from the plate and followed by 500 μL of 1 mmol/L EDTA–PBS per well for 5 min at 37 °C. The dissociated cells were fixed with 2% paraformaldehyde (Fisher Scientific, Fairlawn, NJ, USA) at 4 °C for 30 min, then washed in PBS twice, centrifuged at 1000×g for 5 min, and incubated with 50 μL primary antibody at 37 °C for 1 h. In total, 50 μL of the secondary antibody was added. After incubation for 1 h at 37 °C, the single cell suspension was obtained and analyzed on a FACScan flow cytometer (Beckman Coulter, Fullerton, CA, USA).
Semiquantitative RT–PCR To determine if specific shRNA has a direct effect on inhibiting HSV-1 replication, the level of viral RNA transcripts in the cells was quantified by semiquantitative RT–PCR. Vero cells were grown in 24-well plates. In total, 120 nmol/L shRNA was added prior to virus infection. Every 4 wells were treated by 1 kind of shRNA. HSV-1-infected samples were collected at 12 and 24 h postinfection. The total RNA of Vero cells was extracted by Trizol reagent (Gibco, Grand Island, NY, USA) and quantified by spectrophotometry. The RNA was quantified from 500 ng of total RNA and reverse transcribed to cDNA with random primers (Promega, Madison, WI, USA) using M-MLV reverse transcriptase (Promega, USA) at 42 °C for 45 min. HSV-1 mRNA levels were measured by semiquantitative PCR. The PCR program consisted of an initial 5 min of denaturation at 95 °C and 28 cycles of 30 s at 95 °C, 45 s at 55 °C, and 45 s at 72 °C. β-Actin was amplified simultaneously as the internal control. The sequence of the primers and probes were as follows: HSV-1 gD, 5’-ATCACGGTAGCCCGGCCGTGACA-3’ (sense), 5’-CATACCGGAACGCACCACACAA-3’ (antisense), allowing amplification of a 220 bp product[12], and β-Actin, 5’-TCATCACTATTGGCAACG AGC-3’ (sense), 5’-AACAGCCCGCCTAGAAGCAC-3’ (antisense), generating a product of 399 bp. The PCR products were then separated on a 2% agarose gel containing ethidium bromide. Gels were viewed and photographed under UV illumination using the Fluor-SR multi imager (Amersham Pharmacia Biotech, Uppsala, SE, Sweden). The bands were measured by densitometric analysis. The proportion of the gD gene to the β-Actin gene in densitometries were counted. All reactions were carried out in triplicate.
Statistical analysis All statistical analyses were performed using SPSS13.0 software (SPSS, Chicago, IL, USA). Data were expressed as mean±SD for all experiments. One-way ANOVA was used to assess statistical significance between means. P<0.05 was considered statistically significant.
Results
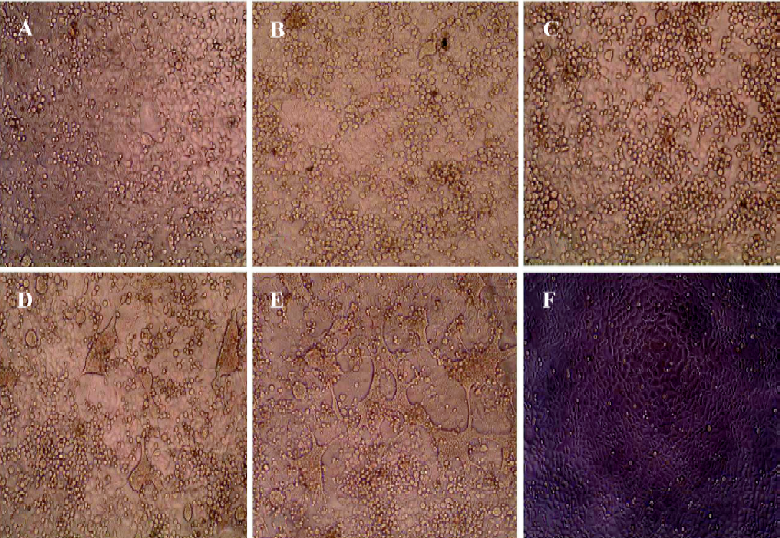
Cytopathic effect inhibited by shRNA in HSV-1-infected Vero cells The efficacy of shRNA in inhibiting HSV-1 replication was first evaluated by transfecting the Vero cells with shRNA-gD1, -gD2, and -gD3 at a final concentration of 30–180 nmol/L. At 24 h post-transfection, the Vero cells were infected with HSV-1 at an MOI of 10. The cytopathic effect (CPE) in infected cells was observed up to 72 h post-infection (hpi). No CPE was seen in the cells treated with 180 nmol/L shRNA-gD1, -gD2, and -gD3 at 48 h postinfection. Silencing results were obtained from the cells treated with 120 nmol/L shRNA. The significant inhibition of CPE was observed at a dose as low as 30 nmol/L of shRNA-gD1 and -gD2. Corresponding to Leishmania CRK1, a proteinkinase was chosen as a negative control. There was no protective effect on the Vero cells treated with shCRK. This indicated that shRNA targeted in the US6 region of HSV-1 genome was able to inhibit the CPE in HSV-1-infected cells (Figure 1).
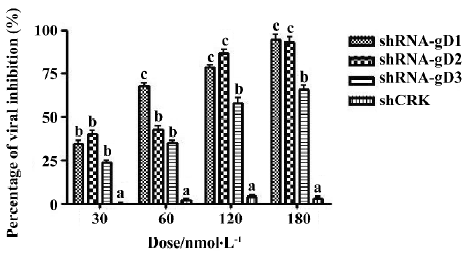
Infectious titer of HSV-1 in shRNA-transfected cells The Vero cells transfected with shRNA-gD1 resulted in the replicative inhibition of HSV-1 by 34.2%±4.3%, 67.1%±5.6%, 78.1%±4.7%, and 95.5%±5.4% (P<0.05 vs shCRK) at concentrations of 30, 60, 120, and 180 nmol/L, respectively (Figure 2). Similar dose-dependent effects were observed when the Vero cells were treated with 30, 60, 120, and 180 nmol/L shRNA-gD2 (39.7%±4.2%, 42.5%±3.6%, 86.3%±5.3%, and 97.3%±2.6%, P<0.05 vs shCRK) and -gD3 (23.5%±2.1%, 34.6%±3.2%, 57.3%±3.7%, and 65.5%±4.9%, P<0.05 vs shCRK). The cells treated with shCRK showed no inhibitory effect on virus replication (P>0.05 vs virus group). The drug concentrations inhibiting 50% and 95% plaque formation were determined by SPSS13.0 (Table 2).

Full table
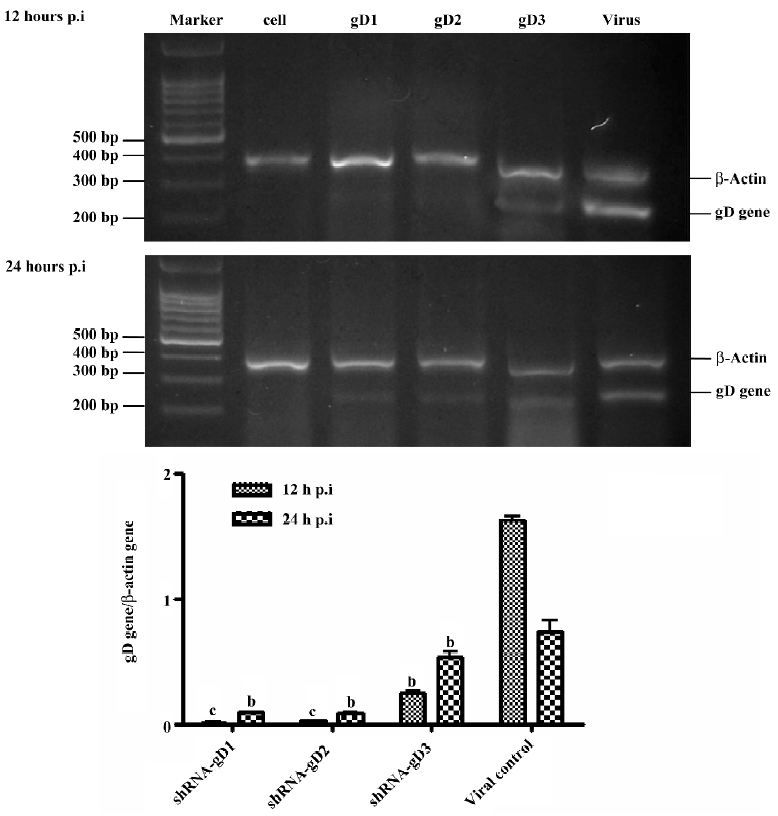
RNA transcripts of HSV-1 in shRNA-transfected cells shRNA was designed to specifically target the HSV-1 US6 gene. Stringent selection of the target region was carried out to ensure effective gene silencing. Vero cells were transfected with 120 nmol/L shRNA-gD1, -gD2, and -gD3, respectively. The data showed that the cells transfected with the 3 shRNA resulted in the inhibition of viral RNA transcription at 12 hpi by 97.42%±2.83%, 96.57%±4.35%, and 85.14%±6.06% (P<0.05 vs virus group), respectively. When the cells were treated with shRNA at 24 hpi, the efficiencies of gD gene inhibition were 86.62%±4.31%, 85.63%±5.56%, and 45.75%±4.55% (P<0.05 vs virus group), respectively (Figure 3). The cells treated with control shCRK showed no effect on the inhibition of viral RNA transcription.
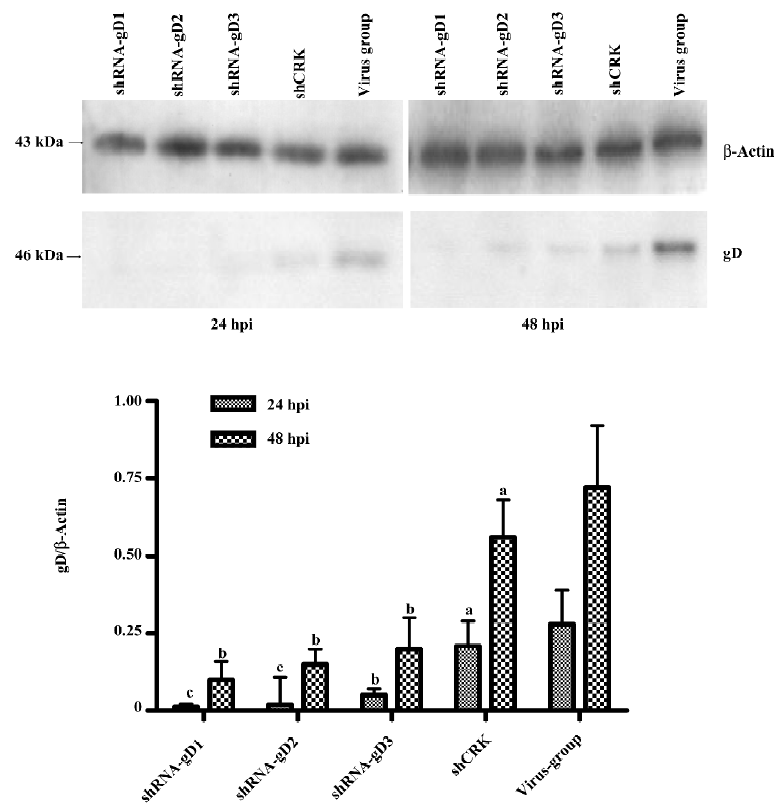
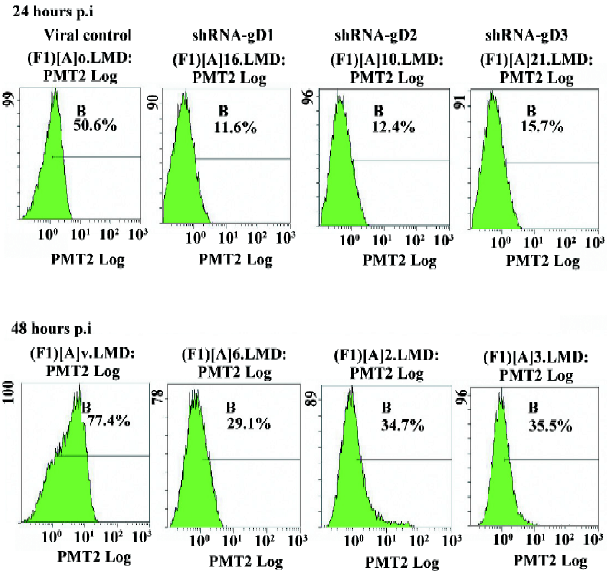
Expression of viral gD in shRNA-transfected cells The expression levels of the gD protein were determined by Western blotting analysis and flow cytometry. A drastic decrease in the amount of gD in shRNA-transfected cells was observed when compared to non-shRNA-transfected cells at 24 hpi (Figure 4). The results of flow cytometry also showed that the expression of gD was significantly reduced in shRNA-treated cells (Figure 5). The reductive rates of the expression of gD in Vero cells transfected with 120 nmol/L shRNA-gD1, -gD2, and -gD3 were 77.8%±3.22%, 76.2%± 4.87%, and 69.7%±3.85% (P<0.05 vs shCRK) at 24 hpi, respectively. At 48 hpi, the efficiencies of gD gene inhibition were 63.3%±3.33%, 55.8%±4.5%, and 54.8%±4.45% (P<0.05 vs shCRK), respectively.
Combination of shRNA-gD1 and -gD2 on the inhibition of the gD expression It has been reported that enhanced antiviral effects of combinations of small interfering RNA (siRNA) have been observed in HIV infection[14]. To evaluate whether cotransfection with 2 specific shRNA targeting different regions of the US6 genome could improve the antiviral effect in this study, a combined final concentration of 120 nmol/L shRNA-gD1 and -gD2 was cotransfected into the Vero cells, followed by infection with HSV-1 as described previously. The inhibitive efficacy of virus replication was measured by plaque assay. The results showed that a similar inhibitory efficacy on HSV-1 replication was observed as compared with shRNA individually used. This suggests that there was no enhanced antiviral effect when shRNA-gD1 and -gD2 were cotransfected.
Discussion
RNAi technology holds great promise in biological and medical fields. The potential of siRNA remains to be established in the area of gene therapy. At the present time, RNAi has been shown to interfere with the replication of a number of different viruses, including HIV[13,14], Coxsackie virus[15], HSV[16], hepatitis B virus[10], and influenza virus[17]. This finding is particularly relevant to infectious disease as it is believed that endogenous RNA interference mechanisms evolved, at least in part, in protecting cells against infectious pathogens. As shRNA-mediated inhibition of virus replication is independent of antigenic variation, immune modulation, and interferon-mediated interference, it has potential application in the therapy and prophylaxis of virus diseases. For therapy, efficiently delivering and expressing siRNA in target cells is necessary.
The inhibition of HSV infection by siRNA has been recently reported[18]. The research focused on genes, such as VP16 and the DNA polymerase genes of HSV-1. The proteins coded by these genes were key enzymes in the replication of herpes viruses. In this research, we chose the US6 gene as the targeting gene. During an infectious cycle, HSV-1 may synthesize more than 70 proteins, which include at least 11 membrane-associated glycosylated proteins[19]. The envelope of HSV-1 contains at least 6 virus-encoded glycoproteins. It has been shown that soluble forms of gD can interact with molecules at the cell surface and interfere with the infection process[20]. HSV-1 entry is composed of several distinct steps[21]. After the initial binding event, virus attachment becomes more stable as gD, and possibly other virus glycoproteins, bind to their cell surface receptor(s)[22,23]. Binding of gD to its entry receptor appears to play 2 roles, the first of which is in securing the attachment event, and the second is in facilitating or initiating penetration of the virus into the cell. Because gD plays such an important role in viral entry, we used it as the target to investigate the effect of RNAi on the inhibition of HSV-1 replication.
If shRNA can inhibit the gD expression, the inhibition of virus penetration is a likely possibility. It was reported that antibodies to gD can alter plaque size and inhibit the formation of polykaryocytes by syncytium-forming mutants[20,21], so we chose to research CPE and use plaque assays to analyze the effects of shRNA in the present study. The efficacy of shRNA was tested by observing CPE and conducting plaque assays at 12, 24, 48, and 72 hpi in preliminary experiments. CPE was observed in all of the groups up to 72 h postinfection. The plaque reduction assay also certificated that there was no significant difference among the shRNA groups and virus-control group.
HSV-1 produced visible plaques on Vero cells at 48 h postinoculation, which developed central clearing as the virus spread outward. The cells transfected with shRNA produced plaques that were characteristically smaller than the control because of the reduced cell-to-cell spread. Microscopy showed that the viral silencing effect of shRNA was specific for HSV-1 and did not disrupt cell morphology. The inhibition of the CPE in Vero cells infected by HSV-1 was observed to last up to 72 hpi. On d 4 of postinfection, no CPE difference was observed in the cells transfected with shRNA as compared to the control. The possible explanation for the slight increase in virus production could be attributed to the fact that transfection of shRNA constructs into Vero cells was not of 100% efficiency. In order to demonstrate the efficiency of shRNA transfection in preliminary experiments, we transfected a control, green fluorescent protein, in Vero cells. We viewed the cells under a 1X-81 microscope (Olympus, Tokyo, Japan) at an excitation wavelength of 480 nm at 12 h post-transfection. Semiquantitative measurements of the percentages of fluorescent cells were counted in each sample of 200 cells taken from 5 randomly-selected fields. The result showed that the percentage of fluorescent cells was 82.4%±4.5%. The efficiency of shRNA transfection was considered the equivalent of the percentages of fluorescent cells[24]. Thus there were approximately 20% cells present in the population that were not transfected with shRNA, but were infected with the virus. These infected cells can produce infectious viruses, masking some of the silencing effects of the transfected cells. Nevertheless, these results indicate that shRNA continues to exert its effect after 24 hpi. The Vero cells transfected with control shRNA (shCRK) had no effect upon viral plaque size following infection. The result indicates that gD gene-specific shRNA is a potent inhibitor of gD synthesis.
In order to investigate the inhibitory effectiveness of using shRNA targeting the HSV-1 gD gene on HSV-1 replication, we used semiquantitative RT–PCR and flow cytometry to detect the viral protein expression and the production of infectious viral particles. The RT–PCR analysis of shRNA-transfected cells showed diminished levels of viral RNA transcripts at 12 and 24 hpi as compared to the control HSV-1-infected cells. To further assess the sustained gene silencing effect of shRNA, virus replication was also determined at 48 hpi by RT–PCR. However, there was an increase in the number of infectious viruses from shRNA-transfected and -infected cells (compared to 12 hpi). An analysis of the viral protein expression also showed a significant reduction in the viral gD protein at 24 and 48 hpi. We also tested the gD expression at 72 h postinfection in preliminary experiment. There was an increase of gD expression observed at 72 hpi, so the effects of inhibition by shRNA could last for at least 48 h.
The results of cotransfection suggested that there was no enhanced antiviral effect when a combination of shRNA-gD1 and -gD2 was used. These particular shRNA probably could not affect the secondary structure of the targets or open more space to other shRNA, or the amount of siRNA-associated proteins was limited for silencing rather than target accessibility[25].
HSV-1 is a human pathogen that infects up to 80% of individuals by adulthood worldwide and causes significant morbidity among immuno-compromised hosts. Unfortunately, few antiviral drugs for HSV-1 are available. The idea that shRNA can be a therapeutic drug is now becoming accepted. Thus the potential of shRNA would be a powerful anti-HSV tool to fight the disease. However, the major concern is how to deliver shRNA to target organs and to determine the length of its effects. Side-effects of treatment are also an issue.
In conclusion, we have shown that sequence-specific US6 shRNA could be used to interfere with viral protein synthesis and to inhibit virus replication. Therefore, they have great potential application for HSV-1 therapy.
Acknowledgements
We would like to thank Mr Ji-lu TANG from the Department of Microbiology, Wuhan University (Wuhan, China) for his kind support and advice on the technical aspects of Western blotting. We are also grateful to Dr Rhea-Beth Markowitz from the Medical College of Georgia (Augusta, GA, USA) for assistance in editing the manuscript.
References
- Lenz G. The RNA interference revolution. Braz J Med Biol Res 2005;38:1749-57.
- Eccleston A, Eggleston AK. RNA interference. Nature 2004;431:337.
- Mello CC, Conte D Jr. Revealing the world of RNA interference. Nature 2004;431:338-42.
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001;411:494-8.
- Gadkari DA. RNA interference and inhibition of viruses. Indian J Med Res 2005;121:147-50.
- Audonnet JC, Winslow J, Allen G, Paoletti E. Equine herpes virus type 1 unique short fragment encodes glycoproteins with homology to herpes simplex virus type 1 gD, gI and gE. J Gen Virol 1990;71:2969-78.
- Paddison PJ, Caudy AA, Bernstein E, Hannon GJ, Conklin DS. Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes Dev 2002;16:948-58.
- Donzé O, Picard D. RNA interference in mammalian cells using siRNAs synthesized with T7 RNA polymerase. Nucleic Acids Res 2002;30:e46.
- Bhuyan PK, Karikò K, Capodici J, Lubinski J, Hook LM, Friedman HM, . Short interfering RNA-mediated inhibition of herpes simplex virus type 1 gene expression and function during infection of human keratinocytes. J Virol 2004; 78: 10 276–81.
- Ren GL, Bai XF, Zhang Y, Chen HM, Huang CX, Wang PZ, et al. Stable inhibition of hepatitis B virus expression and replication by expressed siRNA. Biochem Biophy Res Commun 2005;335:1051-9.
- McSharry JJ. Analysis of virus-infected cells by flow cytometry. Methods 2000;21:249-57.
- Hukkanen V, Rehn T, Kajander R, Sjöroos M, Waris M. Time-resolved fluorometry PCR assay for rapid detection of herpes simplex virus in cerebrospinal fluid. J Clin Microbiol 2000;38:3214-8.
- Ji J, Wernli M, Klimkait T, Erb P. Enhanced gene silencing by application of multiple specific small interfering RNAs. FEBS Lett 2003;552:247-52.
- Novina CD, Murray MF, Dykxhoorn DM, Beresford PJ, Riess J, Lee SK, et al. SiRNA-directed inhibition of HIV-1 infection. Nat Med 2002;8:681-6.
- Yuan J, Cheung PK, Zhang HM, Chau D, Yang D. Inhibition of Coxsackievirus B3 replication by small interfering RNAs requires perfect sequence match in the central region of the viral positive strand. J Virol 2005;79:2151-9.
- Palliser D, Chowdhury D, Wang QY, Lee SJ, Bronson RT, Knipe DM, et al. An siRNA-based microbicide protects mice from lethal herpes simplex virus 2 infection. Nature 2006;439:89-94.
- Hui EK, Yap EM, An DS, Chen IS, Nayak DP. Inhibition of influenza virus matrix (M1) protein expression and virus replication by U6 promoter-driven and lentivirus-mediated delivery of siRNA. J Gen Virol 2004;85:1877-84.
- Zhang YQ, Lai W, Li H, Li G. Inhibition of herpes simplex virus type 1 by small interfering RNA. Clin Exp Dermatol. 2008;33:56-61.
- Spear PG, Longnecker R. Herpesvirus entry: an update. J Virol 2003; 77: 10 179–85.
- Rux AH, Willis SH, Nicola AV, Hou W, Peng C, Lou H, et al. Functional region IV of glycoprotein D from herpes simplex virus modulates glycoprotein binding to the herpesvirus entry mediator. J Virol 1998;72:7091-8.
- Huber MT, Wisner TW, Hegde NR, Goldsmith KA, Rauch DA, Roller RJ, . Herpes simplex virus with highly reduced gD levels can efficiently enter and spread between human keratinocytes. J Virol 2001; 75: 10 309–18.
- Fusco D, Forghieri C, Campadelli-Fiume G. The pro-fusion domain of herpes simplex virus glycoprotein D (gD) interacts with the gD N terminus and is displaced by soluble forms of viral receptors. Proc Natl Acad Sci USA 2005;102:9323-8.
- Zhou G, Ye GJ, Debinski W, Roizman B. Engineered herpes simplex virus 1 is dependent on IL13Ralpha 2 receptor for cell entry and independent of glycoprotein D receptor interaction. Proc Natl Acad Sci USA 2002; 99: 15 124–9.
- Krichevsky AM, Kosik KS. RNAi functions in cultured mammalian neurons. Proc Natl Acad Sci USA 2002; 99: 11 926–9.
- Elbashir SM, Harborth J, Weber K, Tuschl T. Analysis of gene function in somatic mammalian cells using small interfering RNAs. Methods 2002;26:199-213.