
Cardioprotection of sevoflurane postconditioning by activating extracellular signal-regulated kinase 1/2 in isolated rat hearts1
Introduction
Anesthetic-induced postconditioning, currently the latest cardioprotective strategy, like traditional ischemic preconditioning[1], drug-induced preconditioning[2,3], and ischemic postconditioning[4] has evident effects on cardioprotection. Sevoflurane, which is the newest, pre-dominant, and ideal volatile anesthetic, is most commonly used to maintain the state of general anesthesia. It has been reported that sevoflurane-induced postconditioning can protect against ischemic–reperfusion injury[5,6]. However, the mechanism of how sevoflurane postconditioning mediates cardioprotection remains unclear.
The extracellular signal-regulated kinase (ERK)1/2, prototypic members of the mitogen-activated protein kinase family, are the most ideal candidates among the protein kinases that determine the specificity of cellular responses, including cell proliferation, cell differentiation, and cell survival, and are activated by a large variety of extracellular agents[7]. ERK1/2 mediates ischemic[8] and pharmacological[9,10] postconditioning and also plays a critical role in myocardial protection against ischemic–reperfusion injury[11]. This has been demonstrated in previous studies of desflurane-induced preconditioning[12] and isoflurane-induced postconditioning[9]. Whether brief exposure to sevoflurane-induced cardioprotection during early reperfusion is also mediated by ERK1/2 is unknown. The mitochondrion, which is an important cell organ of eukaryon, plays a very critical role in cellular apoptosis and necrosis. Many studies have demonstrated the involvement of mitochondria in cardioprotection[13,14]. Cytochrome c is an important component of the electron transport chain complex of mitochondria, and its release has significant effects on damage induced by ischemic reperfusion by influcing the form of ATP through affecting the electron transfer of the breathing chain. With external stimulation, it is released from mitochondria into the cytosol and contributes to cellular death[15]. Our study aims to research the effect of sevoflurane on heart function, infract size, ERK1/2 activity, mitochondria, and cytochrome c, and discuss the potential mechanism of in cardioprotection and its potential for clinical application.
Materials and methods
Materials Adult, male Sprague–Dawley rats, weighing 250-300 g, were provided by the Experimental Animal Center of Xuzhou Medical College (Xuzhou, China). This study was conducted in accordance with the guidelines of the Animal Care and Use Committee of the University of Xuzhou Medical College. Sevoflurane (lot: 7129) was purchased from Maruishi Pharmaceutical (Fuji Shizuoka, Japan). 2,3,5- Triphenyltetrazolium chloride (TTC) and PD98059 were from Sigma (St Louis, MO, USA). anti-phospho-ERK1/2 (N
Isolated, perfused heart preparation The rats were anaesthetized with an intraperitoneal injection of 40 mg·kg–1 pentobarbital and 500 IU·kg–1heparin. After median sternotomy, the hearts were rapidly isolated and placed in 4 °C Krebs–Henseleit (K-H) buffer, then perfused through the aorta using Langendorff apparatus. The perfusion press was set at 78 mmHg, and the K-H buffer was maintained at 37 °C. The K-H buffer contained (in mmol/L): NaCl 118, KCl 4.7, MgSO4 1.2, KH2PO4 1.2, NaHCO3 25, CaCl2 2.5, glucose 11.1, and EDTA-Na2 0.125 equilibrated with a mixture of 95% oxygen and 5% carbon dioxide (pH, 7.4±0.02).
A saline-filled latex balloon was inserted into the left ventricle through the mitral valve, connected to a pressure transducer (Maclab/4S; AD Instruments, Sydney, NSW, Australia) by a polyethylene catheter, and then connected to a Macintosh 7200/90 computer (Apple, Cupertino, CA, USA) by the Maclab/4S simulated converter. The balloon volume, which remained unchanged throughout the experiment, was adjusted to achieve a left ventricle end-diastolic pressure (LVEDP) of 4-6 mmHg at the beginning of perfusion. The left ventricular function index was monitored continuously by the Macintosh 7200/90 computer: LVEDP, left ventricular developed pressure (LVDP), positive and negative LV dp/dt (dp/dtmax, dp/dtmin), heart rate (HR).
Experimental protocol The whole procedure lasted for 150 min. After equilibration of 30 min, the isolated hearts underwent 30 min of global ischemia followed by 90 min of reperfusion. In total, 3% sevoflurane was administered for 5 min with a 10 min washout. Sevoflurane concentrations were also measured in the buffer by a gas chromatograph (Varian 3400; New York, USA; 0.50±0.04 mmol/L).
Protocol 1 involved the analysis of functional parameters and the determination of infarct size. Our aim was to assess the efficacy of cardioprotection with sevoflurane postconditioning. The rat hearts were randomly divided into 7 groups as follows (n=84, 12 per group): (1) Sham group: perfusion for 150 min in K-H buffer; (2) Control group: global ischemia for 30 min and then 90 min of reperfusion; (3) Post group: four cycles of ischemic postconditioning (25 s of ischemia and 25 s of reperfusion) at the onset of reperfusion after 30 min of ischemia; (4) Sevo group: administration of 3% sevoflurane for 5 min followed by a 10 min washout at the onset of reperfusion after 30 min of ischemia; (5) DMSO group: administration of the PD98059 vehicle DMSO (<0.2%) for 15 min at the onset of reperfusion; (6) PD group: administration of the selective ERK1/2 inhibitor PD98059 (20 μmol/L) in DMSO for 15 min at the onset of reperfusion; and (7) Sevo+PD group: administration of both PD98059 in DMSO and 3% sevoflurane for 15 min at the onset of reperfusion.
Left ventricular hemodynamics and coronary flow (CF) at 30 min of equilibrium were recorded at 30, 60, and 90 min of reperfusion, respectively. Acute infarct size was measured by TTC staining. The configuration of mitochondria was observed by an electron microscope. Western blot analysis was used to determine the contents of cytosolic and mitochondrial cytochrome c at the end of reperfusion
To assess the potential contribution of ERK1/2 in sevoflurane-induced cardioprotection, protocol 2 involved additional hearts being randomly assigned to the abovementioned groups (n=35, 5 per group). After 15 min of reperfusion, the expression of total and phosphorylated forms of ERK1/2 and its downstream target p70S6K was determined by Western blotting.
Determination of myocardial infarct size At the end of the experiment, the hearts were frozen at –20 °C for 2 h and then sliced into 5, 2 mm cross-sections across the long axis. The sections were incubated at 37 °C for 20 min with 1% TTC in 0.1 mol/L phosphate buffer with the pH adjusted to 7.4. Afterwards, the sections were fixed for 10-14 h in 10% formaldehyde and digitally photographed. Tissues that were stained brick red were taken as viable; pale or white tissues were taken as necrotic. A planimetric analysis, using Image Pro-Plus (Media Cybernetics, Silver Spring, USA), was performed to determine infarct size. Because the entire left ventricle was at risk (global ischemia), infarct size was divided by the total area of the myocardium to yield the percentage area of infarction.
Extraction of phospho-ERK (p-ERK) and phospho-p70S6K (p-70S6K) cytosol protein The left ventricular tissue was taken and immediately frozen in liquid nitrogen at –70 °C after 15 min of reperfusion. Subsequently, the tissue was powdered and homogenized in cell lysis buffer for Western blotting. The homogenates were vortexed for 15 s and then centrifuged at 14 000×g for 10 min at 4 °C. The supernatants were used for Western blot analysis as cytosolic proteins. Protein concentrations were determined by the Bradford method using bovine serum albumin as a standard.
Isolation of the subsarcolemmal mitochondria and cytosol protein At the end of reperfusion, the hearts were immediately removed from the perfusion column and placed into buffer A [containing in mmol/L: 100 KCl, 50 3-(morpholino) propanesulfonic acid (MOPS), 1 ethylene-glycol bis tetraacetic acid (EGTA), 5 MgSO4·7H2O, and 1 ATP at pH 7.4] at 4 °C. The cardiac tissue was finely minced, washed twice, and placed in buffer A containing 0.2% bovine serum albumin at 20 mL·g–1 and homogenized with a polytron tissue processor (Brinkman Instruments, Westbury, NY, USA) for 2.5 s at a rheostat setting of 6.0. The homogenate was centrifuged at 584×g for 10 min, the supernatant was saved for the isolation of subsarcolemmal mitochondria (SSM), and the pellet was washed. The combined supernatants were centrifuged at 3015×g for 10 min to sediment SSM. The supernatants were used for Western blot analysis as cytosolic proteins (at a concentration of approximately 2 mg·mL–1). The sediment SSM was suspended in buffer A containing 0.2% bovine serum albumin at 5 mL·g–1 and centrifuged at 3015×g for 10 min again. The sediment SSM was suspended in KME (containing in mmol/L: 100 KCl, 50 MOPS, and 0.5 EGTA) at 2.5 mL·g–1 and centrifuged at 3015×g for 10 min. Finally, the sediment SSM was suspended in KME again (at a concentration of approximately 1.5 mg·mL–1). The equipment and solutions were kept at 4 °C during the isolation process. Mitochondrial and cytosolic protein concentrations were determined by the Bradford method using bovine serum album as a standard.
SSM morphology under the electron microscope The SSM sediment was processed as an electron microscope specimen by fixing in 2.5% glutaral phosphate-buffered saline (PBS), washing with 0.1 mol/L PBS for 30 min, fixing in 1% osmic acid for 2 h, and then washing so that the alcohol and ethyl carbinol gradually dehydrated. The specimens were put in mixed liquor containing acetone and resin (2:1) for 1 h, acetone and resin (1:1) for 1.5 h, and 100% resin for 2 h, and were then embedded with 100% resin and placed in a 37 °C, 45 °C, or 60 °C incubator for 24 h. Finally, the specimens were cut into ultra-thin slices, stuck to a copper screen, and stained with uranyl acetate and lead citrate. Electron pictures were taken randomly at a magnification of ×5000.
Western blot analysis The isolated SSM protein and cytosolic protein samples were mixed with SDS–PAGE sample loading buffer and denatured at 100 °C for 5 min. Equivalent amounts of proteins (50 μg) were separated by SDS–PAGE and transferred to nitrocellulose membranes. The membranes were incubated overnight at 4 °C with the primary antibody, washed 3 times with Tris-buffered saline Tween-20 for 5 min each time, and incubated for 2 h with the secondary antibody conjugated with alkaline phosphatase at room temperature. Immune complexes were detected using a NBT/BCIP assay kit. The scanned images were imported into Adobe Photoshop software (Adobe, San Jose, CA, USA). Scanning densitometry was used for the semiquantitative analysis.
Statistical analysis All values were expressed as mean±SD. A paired t-test was used for comparison within groups over time. The statistical analysis of data between groups was performed by a two-way ANOVA with multiple comparisons or Student’s t-test. Results described as significant are based on a criterion of P<0.05.
Results
Effects on CF and systemic hemodynamics No differences in baseline hemodynamics were observed among the experimental groups (P>0.05). At 30, 60, and 90 min of reperfusion, the other groups, with the exception of the Sham group, had a significant decrease (P<0.05) in +dp/dt, –dp/dt, LVDP, and CF, and a large increase (P<0.05) in LVEDP compared with baseline values. Compared with the Sham group, the other groups had a significant decrease (P<0.05) in +dp/dt, –dp/dt, LVDP and CF, and a large increase (P<0.05) in LVEDP. Compared with the Control group, the Sevo and Post groups had a significant increase (P<0.05) in +dp/dt, -dp/dt, LVDP, and CF, and a large decrease (P<0.05) in LVEDP. There was no significant difference in the Sevo and Post groups (Table 1).
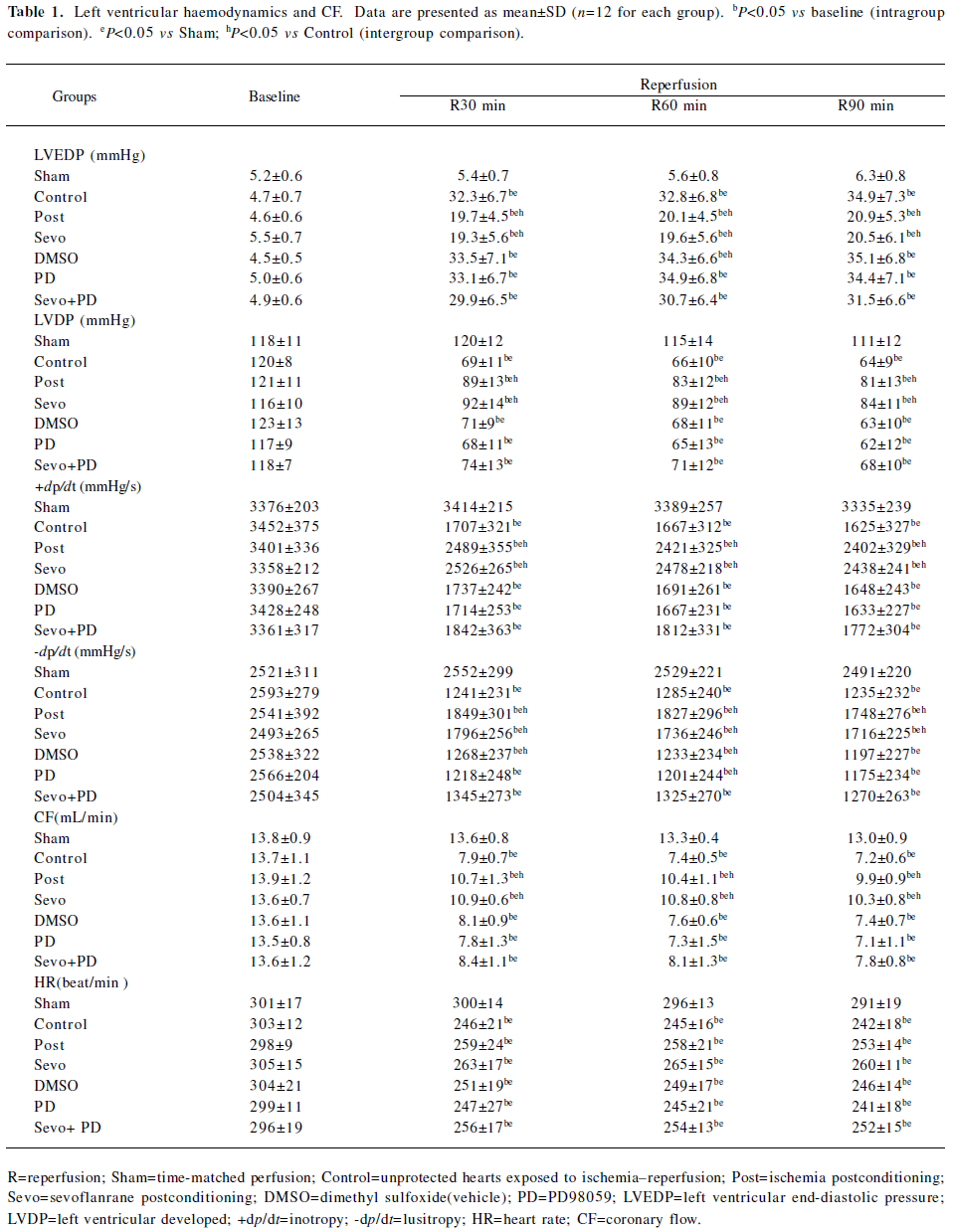
Full table
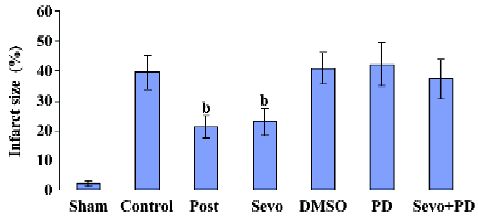
Effects on myocardial infarct size As shown in Figure 1, infarct size in the Sevo and Post groups was significantly reduced (22.9%±4.6% and 21.2%±3.8%, respectively, P<0.05) compared with the Control group (39.4%±5.7%) at the end of reperfusion. Infarct size in the DMSO, PD, and Sevo+PD groups was 40.7%±5.4%, 42.1%±7.2%, and 37.2%±6.7%, respectively, with no statistical differences compared to Control group.
Effects of p-ERK1/2, p-p70S6K, cytochrome c As shown in Figure 2, Western blotting showed the phosphorylated state of ERK1/2 (44 and 42 kDa) and p70S6K (70 kDa) after reperfusion at 15 min, and the content of cytosolic and mitochondrial cytochrome c at the end of reperfusion.
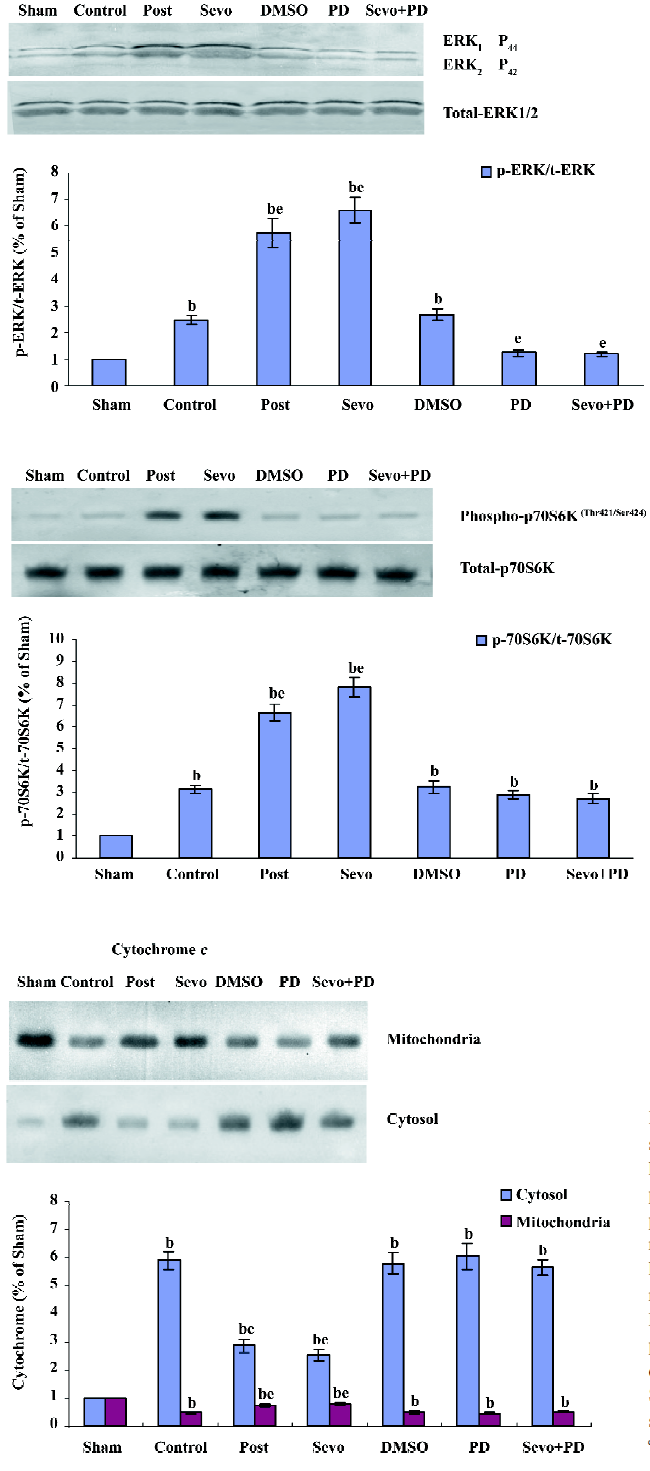
Compared with the sham group, the expression of p-ERK1/2 and p-p70S6K in the control group significantly increased (P<0.05) (Figure 3). Compared with the control group, the expression of p-ERK1/2 and p-p70S6K in the Sevo and Post groups further increased (P<0.05). There were no statistical differences between the Sevo and Post groups, and the DMSO and control groups.
Compared with the sham group, the content of cytochrome c in the other groups significantly increased (P<0.05) in cytosol and obviously decreased (P<0.05) in mitochondria. Compared with the control group, the content of cytochrome c in the Sevo and Post groups significantly decreased (P<0.05) in cytosol and obviously increased (P<0.05) in mitochondria. There were no statistical differences in the Sevo and Post groups.
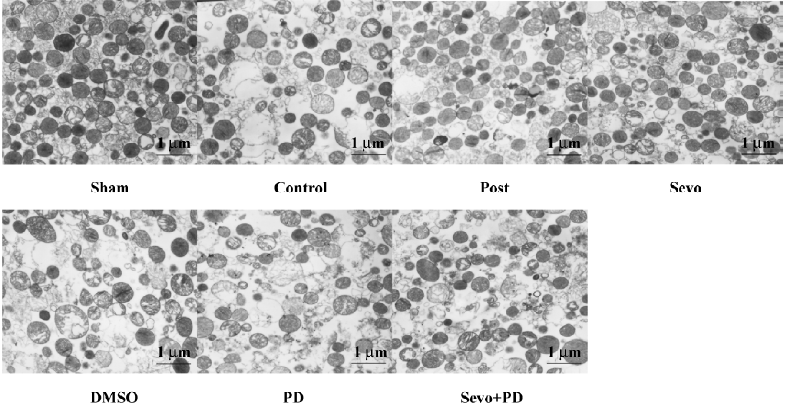
Morphology of SSM with electron microscopy analysis Most isolated SSM in the Sham group exhibited typical morphology. The morphology of mitochondria in the control group exhibited most disruption of the outer membrane, severe swelling of the matrix, decrease of matrix density, and vacuolization of cristae. The Sevo and Post groups had less mitochondria damage, displayed an increase in the quantity of mitochondria, had less swelling of the matrix, and general integrity of membrana external. The DMSO, PD, and Sevo+PD groups were similar to the control group. Magnification was set at ×5000.
Discussion
The model for this experiment is that the ischemic–reperfused myocardium of isolated rat hearts underwent 5 min of sevoflurane-induced postconditioning. The result verified that sevoflurane-induced postconditioning significantly improved +dp/dt, –dp/dt, and LVDP, and at the same time reduced LVEDP in the left artrium of the isolated rat heart after the reperfusion, which indicated that sevoflurane-induced postconditioning could improve contractile and diastolic functions of the ischemic–reperfused myocardium. The large reduction of infract size at the end of reperfusion indicated that sevoflurane-induced postconditioning had a significant effect on cardioprotection.
The mechanism in which sevoflurane postconditioning produces cardioprotection might be similar to that of other volatile anesthetics in inhibiting abnormal activities in the early reperfusion period by its pharmacology or activiating some signaling pathways. With its more favorable physicochemical properties in cardiovascular and autonomic effects, sevoflurane can make the heart rate and hemodynamics more stable and the heart have lower occurrence of arrhythmia and other adverse reactions[16], which may be relevant to the myocardial protection of sevoflurane.
This study confirmed sevoflurane’s ability of improving CF in the myocardium at the period of reperfusion, which has the benefit of improving reflow. The “no reflow” phenomenon in the myocardium is a very important character of reperfusion damage[17]. In the reperfusion period, the capillary in the ischemic zone can not be sufficiently perfused, which causes the decrease of CF. The change of CF can contribute to the development of reperfusion damage. As pharmacology of volatile anesthetics can reduce the resistance of the coronary artery and cause an obvious increase of CF, in some studies, the protection of volatile anesthetics against myocardial damage was found to be relative to the augmentation of CF[18,19]. Sevoflurane can expand the coronary artery. The possibility of “coronary artery steal” is lower than isoflurane, and the inhalation of high concentrations of sevoflurane does not reduce CF. Kehl et al[20] verified that 1.5 MAC sevoflurane did not cause abnormal distribution in dog’s coronary artery, but increased blood flow in coronary artery collateral vessels, and the activation of calcium-activated potassium channels was part of its mechanism. Coronary collateral perfusion is a major determinant of the degree of injury associated with an ischemic event[21]. Harkin et al[22] found that sevoflurane did not affect the coronary artery flow rate in the diastolic phase, and that the expanding effect in the diastolic phase of sevoflurane was different from isoflurane. In our study, sevoflurane significantly increased CF. This may be related to its pharmacological character of decreasing coronary resistance or its protection on lessening the swelling of the myocardial and vascular endothelial cells. These 2 possibilities complement each other to increase the CF and decrease the damage of the myocardial cell, forming non-malignant circulation.
In the present study, by examining Western blot strap, we observed that sevoflurane postconditioning can cause a significant increase of ERK1/2 activity, and PD98059, as an inhibitor of ERK1/2, shut down its activity and offset the protection of sevoflurane postconditioning, which confirmed that ERK1/2 signaling channels play a critical role in cardioprotection induced by sevoflurane postconditioning. In studies of the cardioprotection mechanism, ERK and PI3K were considered as survival kinases pathway of myocardial protection and have been the focus of attention[23]. Coincidentally, the mechanism of preconditioning and postconditioning both include a signal-conducting pathway of the 2 protein kinases. It seems that in the reperfusion period, the 2 forms of myocardial protection may share some signaling pathways that could potentially be common targets of myocardial protection. However, in experiments of different models with different species, ERK- and PI3K-mediated myocardial protection was still questioned.
In 1990, ERK was first described as a serine/threonine protein kinase that was tyrosine phosphorylated by various extracellular signals, including insulin and NGF[24]. Although most studies have verified the role of ERK1/2 as an important, protective signaling pathway both in the setting of preconditioning and postconditioning, it remains controversial. In the setting of ischemic reperfusion induced by a 1.0 MAC isoflurane in vitro rabbit model, Krolikowski et al[25,26] found that at the first 15 min of the reperfusion period, both inhibiting ERK1/2 by PD98059 and inhibiting PI3K by LY294002, abrogated cardioprotection caused by isoflurane postconditioning. They suggested that both pathways play a very important role in cardioprotection induced by isoflurane postconditioning. In an in vitro rabbit model, Darling et al[27] implied that the cardioprotective effect of ischemic postconditioning was dependent on the activation of ERK, but not PI3K, while in an in vitro rats model of ischemic postconditioning, Zhu et al[28] suggested that it relied on the activiation of PI3K, but not ERK1/2. It has been suggested in other studies[29,30,31] that the 2 ways mediate cardioprotection of preconditioning and postconditioning. The differences of these results may depend on the experimental situations and species applied. So far, there are no reports on cardioprotection that are mediated by the ERK1/2 signaling pathway in studies of sevoflurane post-conditioning. In this study, by setting reperfusion through administering 3% sevoflurane in isolated rat hearts, we observed, after 15 min of reperfusion, that the activity of ERK1/2 increased in both the Control and Sevo groups. However, this increase was much more pronounced in the sevoflurane postconditioning group, and PD98059, the inhibitor of ERK1/2, offset the protection induced by sevoflurane postconditioning, which indicated that ERK1/2 was a important signal path in cardioprotection induced by sevoflurane postconditioning. Although ERK1/2 plays a very important role in myocardial protection in our study, the mechanism of mediating myocardial protection is still unclear. The results also require clarification, because we did not specifically examine the biochemical actions of sevoflurane on ERK1/2 phosphorylation, nor did we measure the activities of enzymes in the rat myocardium exposed to ischemia and reperfusion. Nevertheless, our data strongly suggest an important role for ERK1/2 in sevoflurane-induced postconditioning against infarction because the selective inhibition of ERK1/2 blocked myocardial protection induced by sevoflurane during reperfusion. Based on a similar pharmacological character of sevoflurne and isoflurane, we suggest that myocardial protection induced by these 2 ways may have similar signaling pathways, but this needs to be verified by further studies.
The 70 kDa ribosomal protein S6 kinase (p70S6K), the downstream target of ERK1/2, is known to regulate cell growth by inducing protein synthesis components. p70S6K is itself a dual pathway kinase, signaling cell survival as well as growth through differential substrates which include mitochondrial BAD and the ribosomal subunit S6, respectively, and mediates cardioprotection by regulating the activity of apoptosis protein BAD[32]. From our straps, we found that the activity of P70S6K increased when the activity of ERK1/2 increased, and the inhibitor of ERK1/2 can also inhibit the activity of p70S6K. This indicates that sevoflurane postconditioning may mediate cardioprotection by activiating the ERK1/2/p70S6k pathway. However, our study demonstrates sevoflurane postconditioning only can improve the activity of p70S6K. Whether this cardioprotection is mediated by this way still needs subsequent studies.
At 15 min reperfusion, the increase of p-ERK1/2 expression in the Control group, compared with the Sham, indicated that ischemic damage can immediately activate histiocytic ERK1/2, which as an early intracellular signal, might be involved in adjusting stress reactions. Therefore, during ischemia–reperfusion, the course of the increase of ERK1/2 activity may be the reparative process of damaged cell caused by ischemia–reperfusion and may also be a protective mechanism of survival cells, which can protect the myocardium against reperfusion damage by further enhancing the activity of ERK1/2; sevoflurane makes this protection better. Meanwhile, the straps also indicated that ischemic postconditioning, like sevoflurane postconditioning, can reinforce the activity of both ERK1/2 and its downstream target p70S6K, which suggests that ischemic and sevoflurane postconditioning may have similar cardioprotection mechanisms. Our study did not verify if the former is mediated by ERK1/2 like the latter, but it has been demonstrated that the former can significantly increase the activity of ERK1/2 and p70S6K, the protection of which may be relative to this pathway.
In this study, we observed ultrastructure of myocardial mitochondria by using an electron microscope and found that sevoflurane postconditioning can lessen its damage. The mitochondrion, the significant site of energy metabolism, is one of the important organelles of the eukaryotic cell. The mitochondrion was involved in the process of cellular necrosis and apoptosis, is responsible for cellular death way, and may be the common pathway of various protective ways in the ischemic reperfusion period. Cao et al[33] provided unequivocal evidence using a cause–effect approach. They found that the attenuation of mitochondrial Ca2+ overload was responsible for cardioprotection against ischemic–reperfusion-induced injury. Some studies have demonstrated that anesthetics mediate cardioprotection during reperfusion by facilitating the opening of ATP-sensitive potassium (mitoKATP) channels of mitochondria. Obal et al[14] demonstrated that the administration of 5-hydroxydecanoate (5-HD), a specific mitoKATP channel blocker, eliminated the effect of sevoflurane on decreasing infract size of the myocardium, which meant that sevoflurane possibly produced cardioprotection by activating mitoKATP channels. Similarly, Krolikowski et al[34] reported in an in vitro rabbit ischemic–reperfusion model by isoflurane postconditioning, the administration of cyclosporine A, an inhibitor of mPTP, can significantly reduce infract size; however, the administration of mitoKATP inhibitor 5-HD or the mPTP opener atractyloside eliminated this protection, which demonstrated that the protection of isoflurane was mediated by the inhibition of mPTP depending on the activation of mitoKATP channels in vitro. Further research is required to verify if sevoflurane reduces mitochondrial damage by adjusting mitoKATP and whether the effect is direct or indirect.
Cytochrome c has a double function: (1) it is involved in electron transfer; and (2) it is an important initiator of apoptosis. Several studies have demonstrated that the release of cytochrome c matters more than the deprivation of mitochondrial respiration activity in the process of apoptosis[15]. The release of cytochrome c from the outer mitochondrial membrane triggered the activity of caspase-3 and formed the cascades that evoked cellular death[35]. Ischemic–reperfusion damage may lead to the release of cytochrome c from mitochondria into cytosol, cause functional impairment of mitochondria, and contribute to cellular death[36,37]. Qian et al[2] reported that isoflurane preconditioning protects against ischemic injury partly by reducing cytochrome c release from subsarcolemmal mitochondria in vitro. In our study, we found that sevoflurane postconditioning can diminish the release of cytochrome c from mitochondria into cytosol. This may be closely correlated to the decrease of myocardial infract size. So far, there are no reports on the mechanism of how sevoflurane postconditioning regulates the release of cytochrome c. We suggest that sevoflurane affects some agents in cytosol, or activates signaling pathways by decreasing mitochondrial damage. Recent studies have indicated that the inhibition of the release of cytochrome c has an important effect on the damaged myocardium, but the reason why cytochrome c is released from mitochondria and other regulatory mechanisms are still unclear.
From the straps, we found that the release of cytochrome c in the downstream of ERK1/2 increased in mitochondria along with the increase of the activity of ERK1/2; however, the inhibitor of ERK1/2 offset this effect, which indicates that sevoflurane postconditioning possibly mediates myocardial preservation by activiating the anti-apoptotic signaling pathway of ERK1/2/cytochrome c. The present study confirms that sevoflurane postconditioning can lessen the release of cytochrome c from mitochondria. Whether its myocardial preservation is brought by this pathway still needs to be verified by further studies.
The present study demonstrates the effect of cardio-protection of sevoflurane postconditioning, which is similar to other pharmacological postconditioning. There are numerous experimental studies demonstrating that both pharmacological preconditioning[38–40] and postconditioning[41–43] have cardioprotective effects. There is no relevant report comparing either of the protective approaches. However, despite nearly 20 years of research in the field of pharmacological preconditioning and the effectiveness of pharmacological preconditioning for protecting the ischemic myocardium, there are no preconditioning-based therapies that are routinely used in clinical medicine at the current time. Part of the problem is the need to administer therapy prior to the known ischemic event. Pharmacological postconditioning is used as an accompaniment with reperfusion after ischemia, and is a relatively novel strategy of cardioprotection associated with reperfusion of the myocardium. It is more likely than preconditioning to be feasible as a clinical application to patients undergoing acute myocardial ischemic disease. So far, the efficacy of these new approaches has been proven preclinically using experimental models, but the actual mechanism of protection remains unclear and their real clinical relevance and ability to reduce human myocardial infarction-associated morbidity and mortality remains to be determined.
In summary, the current results confirm that 3% sevoflurane-induced postconditioning reduces myocardial infarct size and improves construction and relaxation of the myocardium in isolated Sprague–Dawley rat hearts with acute myocardial ischemic–reperfusion injury. A selective pharmacological inhibitor of ERK1/2 abolished the cardiopro-tection of sevoflurane-induced postconditioning, which revealed that the effect of the cardioprotection of sevoflurane postconditioning was mediated by ERK1/2 signaling channels. In addition, sevoflurane-induced postconditioning attenuated mitochondrial dysfunction, and inhibited the release of cytochrome c from mitochondria, which was in part mediated by ERK1/2 signaling channels. The cardioprotective mechanisms of inhalation anesthetics like sevoflurane are complicated and numerous, and require further exploration.
References
- Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 1986;74:1124-36.
- Qian LP, Zhu SS, Cao JL, Zeng YM. Isoflurane preconditioning protects against ischemia-reperfusion injury partly by attenuating cytochrome c release from subsarcolemmal mitochondria in isolated rat hearts. Acta Pharmacol Sin 2005;26:813-20.
- Zhang Y, Chen ZW, Girwin M, Wong TM. Remifentanil mimics cardioprotective effect of ischemic preconditioning via protein kinase C activation in open chest of rats. Acta Pharmacol Sin 2005;26:546-50.
- Zhao ZQ, Corvera JS, Halkos ME, Kerendi F, Wang NP, Guyton RA, et al. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning. Am J Physiol Heart Circ Physiol 2003;285:H579-88.
- He W, Zhang FJ, Wang SP, Chen G, Chen CC, Yan M. Postconditioning of sevoflurane and propofol is associated with mitochondrial permeability transition pore. J Zhejiang Univ Sci B 2008;9:100-8.
- Deyhimy DI, Fleming NW, Brodkin IG, Liu H. Anesthetic preconditioning combined with postconditioning offers no additional benefit over preconditioning or postconditioning alone. Anesth Analg 2007;105:316-24.
- Torii S, Nakayama K, Yamamoto T, Nishida E. Regulatory mechanisms and function of ERK MAP kinases. J Biochem 2004;136:557-61.
- Darling CE, Jiang R, Maynard M, Whittaker P, Vinten-Johansen J, Przyklenk K. Postconditioning via stuttering reperfusion limits myocardial infarct size in rabbit hearts: role of ERK1/2. Am J Physiol Heart Circ Physiol 2005;289:H1618-26.
- Krolikowski JG, Weihrauch D, Bienengraeber M, Kersten JR, Warltier DC, Pagel PS. Role of Erk1/2, p70s6K, and eNOS in isoflurane-induced cardioprotection during early reperfusion in vitro. Can J Anaesth 2006;53:174-82.
- Yang XM, Krieg T, Cui L, Downey JM, Cohen MV. NECA and bradykinin at reperfusion reduce infarction in rabbit hearts by signaling through PI3K, ERK, and NO. J Mol Cell Cardiol 2004;36:411-21.
- Hausenloy DJ, Yellon DM. Reperfusion injury salvage kinase signalling: taking a RISK for cardioprotection. Heart Fail Rev 2007;12:217-34.
- Toma O, Weber NC, Wolter JI, Obal D, Preckel B, Schlack W. Desflurane preconditioning induces time-dependent activation of protein kinase C epsilon and extracellular signal-regulated kinase 1 and 2 in the rat heart in vitro. Anesthesiology 2004;101:1372-80.
- Chiu PY, Leung HY, Siu AH, Poon MK, Ko KM. Schisandrin B decreases the sensitivity of mitochondria to calcium ion-induced permeability transition and protects against ischemia-reperfusion injury in rat hearts. Acta Pharmacol Sin 2007;28:1559-65.
- Obal D, Dettwiler S, Favoccia C, Scharbatke H, Preckel B, Schlack W. The influence of mitochondrial KATP-channels in the cardioprotection of preconditioning and postconditioning by sevoflurane in the rat in vitro. Anesth Analg 2005;101:1252-60.
- Jiang X, Wang X. Cytochrome c-mediated apoptosis. Annu Rev Biochem 2004;73:87-106.
- Ebert TJ. Cardiovascular and autonomic effects of sevoflurane. Acta Anaesthesiol Belg 1996;47:15-21.
- Kloner RA, Ganote CE, Jennings RB. The “no-reflow” phenomenon after temporary coronary occlusion in the dog. J Clin Invest 1974;54:1496-508.
- Kevin LG, Novalija E, Riess ML, Camara AK, Rhodes SS, Stowe DF. Sevoflurane exposure generates superoxide but leads to decreased superoxide during ischemia and reperfusion in isolated hearts. Anesth Analg 2003;96:949-55.
- Feng J, Fischer G, Lucchinetti E, Zhu M, Bestmann L, Jegger D, et al. Infarct-remodeled myocardium is receptive to protection by isoflurane postconditioning: role of protein kinase B/Akt signaling. Anesthesiology 2006;104:1004-14.
- Kehl F, Krolikowski JG, Tessmer JP, Pagel PS, Warltier DC, Kersten JR. Increases in coronary collateral blood flow produced by sevoflurane are mediated by calcium-activated potassium (BKCa) channels in vitro. Anesthesiology 2002;97:725-31.
- Kersten JR, Toller WG, Gross ER, Pagel PS, Warltier DC. Diabetes abolishes ischemic preconditioning: role of glucose, insulin, and osmolality. Am J Physiol Heart Circ Physiol 2000;278:H1218-24.
- Harkin CP, Pagel PS, Kersten JR, Hettrick DA, Warltier DC. Direct negative inotropic and lusitropic effects of sevoflurane. Anesthesiology 1994;81:156-67.
- Hausenloy DJ, Yellon DM. Survival kinases in ischemic preconditioning and postconditioning. Cardiovasc Res 2006;70:240-53.
- Boulton TG, Yancopoulos GD, Gregory JS, Slaughter C, Moomaw C, Hsu J, et al. An insulin-stimulated protein kinase similar to yeast kinases involved in cell cycle control. Science 1990;249:64-7.
- Krolikowski JG, Weihrauch D, Bienengraeber M, Kersten JR, Warltier DC, Pagel PS. Role of Erk1/2, p70s6K, and eNOS in isoflurane-induced cardioprotection during early reperfusion in vitro. Can J Anaesth 2006;53:174-82.
- Chiari PC, Bienengraeber MW, Pagel PS, Krolikowski JG, Kersten JR, Warltier DC. Isoflurane protects against myocardial infarction during early reperfusion by activation of phosphatidylinositol-3-kinase signal transduction: evidence for anesthetic-induced postconditioning in rabbits. Anesthesiology 2005;102:102-9.
- Darling CE, Jiang R, Maynard M, Whittaker P, Vinten-Johansen J, Przyklenk K. Postconditioning via stuttering reperfusion limits myocardial infarct size in rabbit hearts: role of ERK1/2. Am J Physiol Heart Circ Physiol 2005;289:H1618-26.
- Zhu M, Feng J, Lucchinetti E, Fischer G, Xu L, Pedrazzini T, et al. Ischemic postconditioning protects remodeled myocardium via the PI3K-PKB/Akt reperfusion injury salvage kinase pathway. Cardiovasc Res 2006;72:152-62.
- Hausenloy DJ, Tsang A, Mocanu MM, Yellon DM. Ischemic preconditioning protects by activating prosurvival kinases at reperfusion. Am J Physiol Heart Circ Physiol 2005;288:H971-6.
- Yang XM, Proctor JB, Cui L, Krieg T, Downey JM, Cohen MV. Multiple, brief coronary occlusions during early reperfusion protect rabbit hearts by targeting cell signaling pathways. J Am Coll Cardiol 2004;44:1103-10.
- Tsang A, Hausenloy DJ, Mocanu MM, Yellon DM. Postconditioning: a form of “modified reperfusion” protects the myocardium by activating the phosphatidylinositol 3-kinase-Akt pathway. Circ Res 2004;95:230-2.
- Harada H, Andersen JS, Mann M, Terada N, Korsmeyer SJ. p70S6 kinase signals cell survival as well as growth, inactivating the pro-apoptotic molecule BAD. Proc Natl Acad Sci USA 2001;98:9666-70.
- Cao CM, Yan WY, Liu J, Kam KW, Zhan SZ, Sham JS, et al. Attenuation of mitochondrial, but not cytosolic, Ca2+ overload reduces myocardial injury induced by ischemia and reperfusion. Acta Pharmacol Sin 2006;27:911-8.
- Krolikowski JG, Bienengraeber M, Weihrauch D, Warltier DC, Kersten JR, Pagel PS. Inhibition of mitochondrial permeability transition enhances isoflurane-induced cardioprotection during early reperfusion: the role of mitochondrial KATP channels. Anesth Analg 2005;101:1590-6.
- Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S. A caspase–activated Dnase that degardes DNA during apoptosis, and its inhibitor ICAD. Nature 1998;391:43-50.
- Czerski LW, Szweda PA, Szweda LI. Dissociation of cytochrome c from the inner mitochondrial membrane during cardiac ischemia. J Biol Chem 2003;278:34499-504.
- Hu XM, Zhang Y, Zeng FD. Effects of beta-aescin on apoptosis induced by transient focal cerebral ischemia in rats. Acta Pharmacol Sin 2004;25:1267-75.
- Jones SP, Zachara NE, Ngoh GA, Hill BG, Teshima Y, Bhatnagar A, et al. Cardioprotection by N-acetylglucosamine linkage to cellular proteins. Circulation 2008;117:1172-82.
- Kaneda K, Miyamae M, Sugioka S, Okusa C, Inamura Y, Domae N, et al. Sevoflurane enhances ethanol-induced cardiac preconditioning through modulation of protein kinase C, mitochondrial KATP channels, and nitric oxide synthase, in guinea pig hearts. Anesth Analg 2008;106:9-16.
- Lai IR, Chang KJ, Tsai HW, Chen CF. pharmacological preconditioning with simvastatin protects liver from ischemia–reperfusion injury by heme oxygenase-1 induction. Transplantation 2008;85:732-8.
- Huhn R, Heinen A, Weber NC, Hollmann MW, Schlack W, Preckel B. Hyperglycaemia blocks sevoflurane-induced postconditioning in the rat heart in vivo: cardioprotection can be restored by blocking the mitochondrial permeability transition pore. Br J Anaesth 2008;100:465-71.
- Baxter GF, Burley DS. Reperfusion and calculated RISKs: pharmacological postconditioning of human myocardium. Br J Pharmacol 2008;153:1-3.
- Zang WJ, Sun L, Yu XJ. Cardioprotection of ischemic postcon-ditioning and pharmacological post-treatment with adenosine or acetylcholine. Sheng Li Xue Bao 2007;59:593-600.