
Electropharmacological properties of telmisartan in blocking hKv1.5 and HERG potassium channels expressed on Xenopus laevis oocytes1
Introduction
Telmisartan, a specific, non-peptide, orally-active angiotensin II type-1 (AT1) receptor blocker (ARB), is the newest available drug class for the treatment of hypertension[1]. Telmisartan has a unique aromatic group that is modified so that the drug has good liposolubility and tissue penetrativity. Simultaneously, its AT1 binding affinity is greater than other selective AT1 receptor blockers, and in functional and ligand binding experiments, it produces a long-lasting competitive antagonism because of its different aromatic group[2].
Angiotensin II and aldosterone produce pro-arrhythmic effects by several mechanisms, including the modulation of voltage-dependent potassium (K+) channels involved in human cardiac repolarization[3]. Drugs that inhibit the renin–angiotensin–aldosterone system exert anti-arrhythmic actions that are related to the blockade of the pro-arrhythmic actions of angiotensin II and aldosterone. These anti-arrhythmic actions include the inhibition of electrical and structural cardiac remodeling, inhibition of neurohumoral activation, reduction of blood pressure, and stabilization of electrolyte disturbances. Several voltage-dependent outward K+ currents play a critical role in repolarization, and thus determine the human cardiac action potential duration[4,5]. The Shaker-related hKv1.5 channel has been cloned from human ventricles[6] and has been identified as the counterpart of the Ikur described in human atrial myocytes[7]. The expression of the human ether-a-go-go-related gene (HERG), identified as the locus of congenital long QT syndrome (LQTS, is a heart condition associated with prolongation of repolarization following depolarization of the cardiac ventricles) type 2 mutations, reveals inwardly-rectifying K+ currents similar to IKr suggesting that HERG underlies cardiac IKr[8] even when functional IKr channels result from the co-assembly of HERG α-subunits and MinK-related peptide β-subunits[9]. Several angiotensin II AT1 receptor antagonists (candesartan, E3174, eprosartan, irbesartan, and losartan) and aldosterone receptor antagonists (canrenoic acid and spironolactone) that directly modulate the activity of the voltage-dependent K+ channels have been reviewed. Recently, it has been described that losartan, the prototype of the AT1 receptor antagonists, and its active metabolite, E3174, at clinically relevant concentrations, directly modify delayed rectifier K+ currents involved in human cardiac repolarization[10]. Losartan inhibits hKv1.5 and HERG currents, whereas E3174 inhibits hKv1.5 and enhances HERG currents. Moreover, these actions are correlated with modifications on the action potential duration. These findings suggested that the AT1 receptor antagonists might exert different effects on K+ currents responsible for repolarization.
However, the effects of telmisartan on cardiac K+ channels have been unknown until now. Therefore, the present study was undertaken to analyze the biophysical properties of telmisartan on hKv1.5 and HERG cloned from a human heart. The results indicated that telmisartan blocked hKv1.5 at plasma concentration levels of therapeutic doses, whereas the blockade of HERG channels occurred only at supra plasma concentration levels of therapeutic doses.
Materials and methods
Molecular biology The human Kv1.5 gene (a kind gift from Dr Maria L GARCIA, Merck & Co, Inc, USA) was subcloned into a pCI-neo vector. HERG was subcloned into the pSP64 plasmid expression vector (Promega, Madison, WI, USA), which was a kind gift from Professor Michael C SANGUINETTI (University of Utah, Salt Lake City, Utah,USA). Before use in experiments, each construct was confirmed with restriction mapping and DNA sequencing of the PCR-amplified segment. Complementary RNA for injection into oocytes were prepared with a T7 mMESSAGE mMACHINE kit (Ambion, Austin, TX, USA) and SP6 Cap-Scribe (Roche Molecular Biochemicals, Mannheim, Germany) after linearization of the expression construct with XhoI and EcoRI, respectively.
Isolation and injection of oocytes The isolation and injection of oocytes were described in a previous study[11]. Xenopus frogs were anesthetized by immersion in 0.2% tricaine for 15−30 min. Ovarian lobes were digested with 1.5 g/L type 1A collagenease (Sigma, St Louis, MO, USA) in Ca2+-free ND96 solution for 1 h to remove follicle cells. Stage IV and V Xenopus laevis oocytes was injected with 46 nL of HERG or hKv1.5 cRNA and then cultured in ND96 solution supplemented with 100 U/L penicillin, 100 U/L streptomycin, and 2.5 mmol/L pyruvate at 18 °C for 1–3 d before use in the voltage clamp experiments. The ND96 solution contained (in mmol/L) 96 NaCl, 2 KCl, 1.8 CaCl2, 2 MgCl2, and 5 HEPES. The pH was adjusted to 7.6 with NaOH.
Two-microelectrode voltage clamp of oocytes A TEV-200 amplifier (Dagan, Minneapolis, Minnesota, USA) and a standard 2-microelectrode voltage clamp technique were used to record currents. Currents were recorded at room temperature (22–24 °C) 2–10 d after cRNA injection. Glass microelectrodes were filled with 3 mol/L KCl (internal recording solution), and their tips were broken to obtain resistances of 1–3 megohms. pCLAMP software (version 9.2; Molecular Devices, Union City, CA, USA) and a 1322A interface (Molecular Devices, Union City, CA, USA) were used to generate voltage commands. Currents were digitally sampled at 5 kHz and filtered at 2 kHz. Leak and capacitive currents were not corrected. The oocyte was superfused with ND96 solution at a rate of 1 mL/min, and the membrane potential was held at –80 mV between test pulses, which were applied at a rate of 1−3/min. Currents were measured before and 10 min after drug application to the bath.
Drugs Telmisartan (Sigma, USA) was prepared as a 10 mmol/L stock solution. Before the experiments, the stock solution was diluted with external solution to reach the desired final concentration. Oocytes were exposed to telmisartan solutions until steady-state effects were achieved, usually within 15 min.
Data analyses Clampfit 9.2 software (Molecular Devices, USA) was employed for the data collection and analyses. Origin 6.0 software (nonlinear curve fitting; Origin Lab Corporation, Northampton, MA, USA) was used to fit the data, calculate time constants, and plot histograms. Fractional blockade was defined as follows: f=1–Idrug/Icon, where Icon and Idrug are the current amplitudes in the absence and the presence of telmisartan, respectively. Dose–response curves were fit by the Hill equation as follows:
(Icon–Idrug)/Icon=Bmax/(1+[IC50/D]n), where Bmax is the maximum blockade of currents, IC50 is the concentration of telmisartan for a half-maximum blockade; D is the concentration of telmisartan, and n is the Hill coefficient. The activation curve was approximated by the normalized conductance–voltage relationship. The activation and inactivation curves were fitted with a Boltzmann distribution:
y=A/(1+exp[{V1/2–Vm}/k]),
where A is the amplitude term, V1/2 is the midpoint of activation or inactivation, Vm is the test potential, and k represents the slope factor of the curve. To describe the time course of current activation, inactivation, and/or deactivation upon depolarization, as well as the tail currents upon repolarization, an exponential analysis was used as an operational approach, fitting the current traces to the following equation:
y =C+ A1exp (–t/τ1)+A2exp (–t/τ2)+ . . .+Anexp (–t/τn),
where τ1, τ2, and τn are the system time constants; A1, A2, and An are the amplitudes of each component of the exponential; and C is the baseline value. The curve-fitting procedure used a non-linear least squares (Gauss–Newton) algorithm. The results were displayed in linear and semilogarithmic format, together with the difference plot. Goodness of fit was judged by the χ2 criterion and by inspection for systematic, non-random trends in the difference plot. Data are presented as mean±SEM. Student’s t-tests for paired and unpaired data were used to compare control conditions with intervention factors. P<0.05 was considered to be statistically significant.
Results
Effects of telmisartan on hKv1.5 currents
Voltage- and concentration-dependent blockade of hKv1.5 current by telmisartan in Xenopus oocytes The membrane potential was held at –80 mV. Figure 1A shows hKv1.5 superimposed current traces recorded by applying 1.5 s pulses from –60 to +60 mV in a 10 mV step, in the absence and presence of 1 µmol/L telmisartan. Outward currents were followed by decaying tail currents upon repolarization to –30 mV. Figure 1B and 1C shows the current–voltage relationship of the peak currents and 1.5 s end-pulse currents obtained in the absence and presence of 1 µmol/L telmisartan. Telmisartan significantly decreased the 1.5 s end-pulse current amplitude at potentials positive to –10 mV (n=6, P<0.05); however, it only slightly diminished the peak current amplitude. As shown in Figure 1D, the blockade ratios of 1.5 s end-pulse currents were 0.1 at –20 mV, 0.33 at –10 mV, 0.44 at 0 mV, 0.55 at +10 mV, and 0.53 at +60 mV, respectively; however the blockade ratio of peak currents was only 0.08 at +60 mV, which indicated the blockade was voltage-dependent.
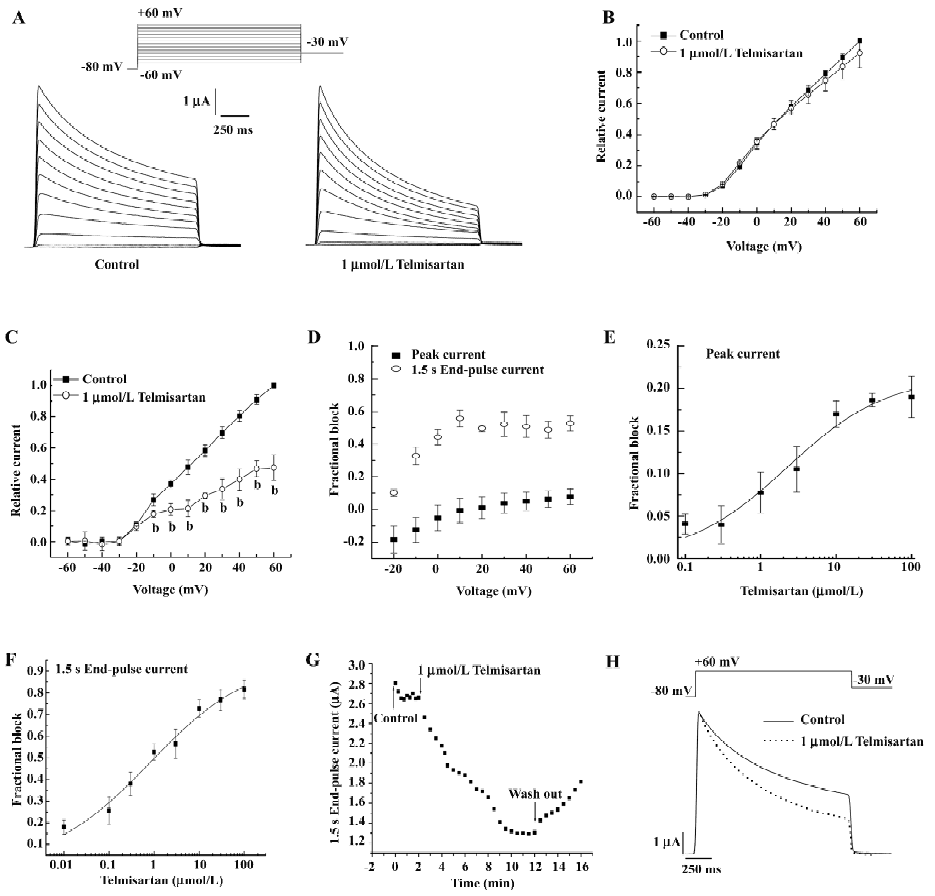
In Figure 1E, the concentration-dependence of telmisartan-induced inhibition was measured on peak Kv1.5 current to +60 mV. Surprisingly, at concentrations between 0.1 and 10 µmol/L, the blockade increased progressively as the concentration of the drug was augmented; however, at 30 and 100 µmol/L, telmisartan induced a similar amount of blockade (approximately 20%). Fitting the concentration–response data to the Hill function, an IC50 (half-maximal inhibition concentration) value of 2.25±0.97 µmol/L with a Bmax of 21.48%±2.04% was obtained. Figure 1F shows the concentration–response curve derived from the reduction of the 1.5 s end-pulses to +60 mV. In this case, the value of IC50 and the Bmax averaged 0.82±0.39 µmol/L and 95.74%±6.98%, respectively. Meanwhile, the blockade at the 1.5 s end-pulse currents was partially reversible (Figure 1G), and control currents were elicited with the application of depolarizing step pulses from a holding potential of –80 mV to a test potential of +40 mV, lasting 10 min to a steady state. Telmisartan was then applied at a concentration of 1 µmol/L to the bath for 10 min, rapidly increased blockade was shown in successive pulses. The inhibitory effect of telmisartan was slowly and partially reversible with washout.
Effect of telmisartan on the inactivation of hKv1.5 channels in Xenopus oocytes As shown in Figure 1H, superimposed current traces were obtained by applying 1.5 s pulses to +60 mV and then returned to –30 mV before and after blockade by 1 µmol/L telmisartan. In control conditions, 1 component was required to describe the time course of the slow and partial inactivation of the channels (693.74±23.16 ms, n=5). However, in the presence of telmisartan, the currents were better fitted to a biexponential function. The time constant of the fast falling phase was considered as the τ (19.56±0.46 ms), whereas the slow time constant reflected the inactivation (523.85±10.28 ms, n=5, P<0.05 vs control conditions). Telmisartan accelerated the inactivation of the channels. These actions of telmisartan are suggestive of an open-channel block mechanism.
Effects of telmisartan on HERG currents
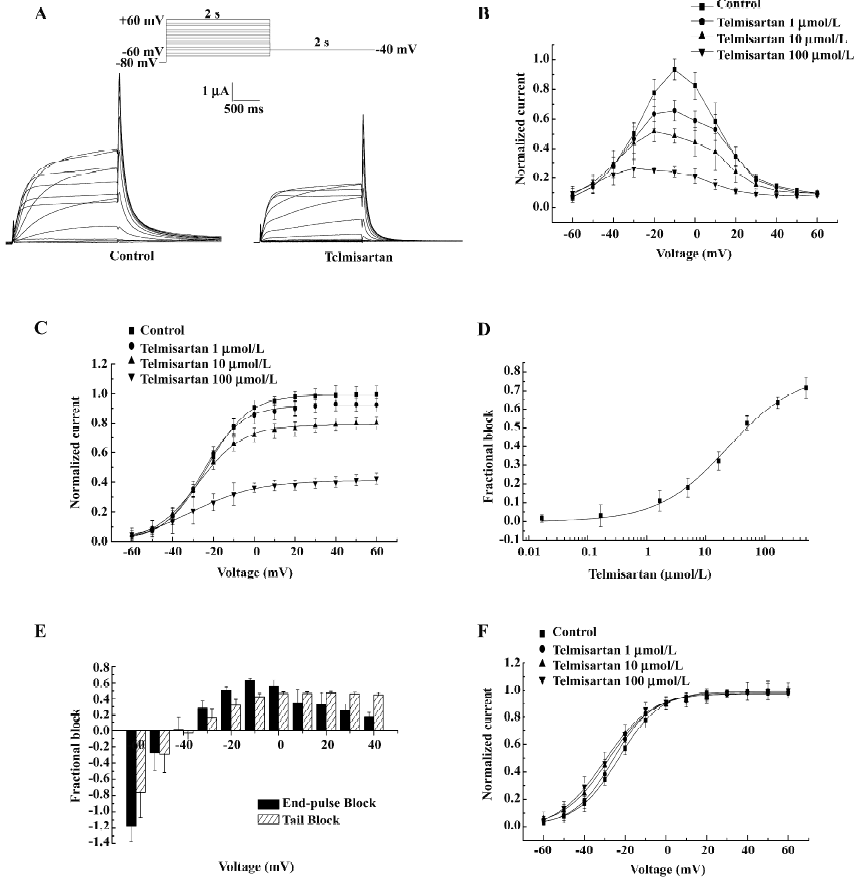
Concentration- and voltage-dependent blockade of HERG currents by telmisartan in Xenopus oocytes Currents were elicited by 2 s depolarizing pulses to potentials ranging from –60 to +60 mV. An example of HERG currents recorded before and after the addition of 50 µmol/L telmisartan is shown (Figure 2A). The normalized I–V (current-voltage) relationship of HERG channels for step currents measured at the end of the 2 s pulses (Figure 2B) and the tail currents measured on return to –40 mV (Figure 2C) were reduced in a concentration-dependent manner by telmisartan. The value of IC50 was 24.35±5.06 µmol/L (n=6) at 0 mV (Figure 2D). Blockade was also voltage-dependent. At the more negative test potential, telmisartan exerted comparatively little effect on IHERG, and in some experiments, the tail current magnitude was greater in the presence of the drug than in control solution. At test potentials greater than –40 mV, a marked inhibitory effect was observed in all cells for step currents and peak tail currents. Furthermore, greater blockade was apparent at more depolarized potentials (Figure 2B,2C). Figure 2E represents the fractional blockade of HERG channel step currents and tail currents at different test potentials. At voltages ranging from –40 to +40 mV, the fractional blockade of the tail currents gradually increased and reached a steady state at 0 mV, with values of –0.02 at –40 mV, 0.15 at –30 mV, 0.31 at –20 mV, 0.42 at –10 mV, 0.46 at 0 mV, and 0.45 at +20 mV, respectively. However, the fractional blockade of the 2 s end-pulse currents first increased from –40 mV to –10 mV following a gradual decrease from –10 mV to +40 mV, with values of 0.01 at –40 mV, 0.28 at –30 mV, 0.50 at –20 mV, 0.62 at –10 mV, 0.55 at 0 mV, and 0.17 at +40 mV, respectively. The difference of blockade may be attributed to different gating mechanisms. In addition, the peaks of the I–V relationship for HERG channels were shifted to the left after using telmisartan, suggesting a negative shift in the voltage dependence of activation. This was confirmed by a tail current analysis (Figure 2F). For HERG channels, the half-point activation value was –22.91±0.71 mV (control) versus –25.83±0.71 mV (telmisartan) at 1 µmol/L, –28.20±0.91 mV at 10 µmol/L and –29.95±1.03 mV at 100 µmol/L. Telmisartan shifted the voltage dependence of the HERG channel activation curve by –2.92±0.01 mV at 1 µmol/L, –5.29±0.20 mV at 1 µmol/L, and –7.04±0.28 mV at 100 µmol/L (P<0.05, n=6), with no significant change in the slope factors [10.28±0.61 mV (control) vs 9.67±0.59 mV at 1 µmol/L, 11.17±0.73 mV at 10 µmol/L, and 11.85±0.79 mV at 100 µmol/L for telmisartan, respectively, P>0.05, n=6]. Thus the dual effect of telmisartan on the IHERG tail magnitude may be explained by both the drug inhibition on IHERG and by producing a leftward shift in the voltage-dependent activation of the currents.
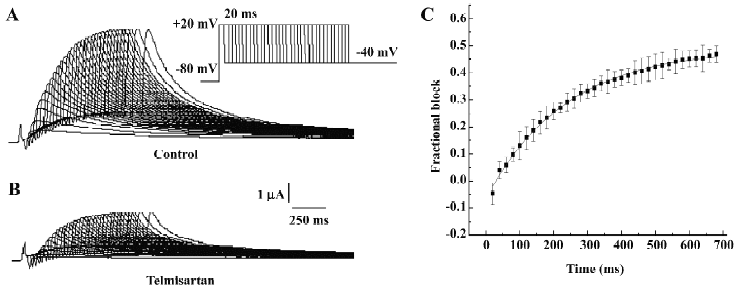
Time-dependent blockade of HERG currents by telmisartan in Xenopus oocytes The time course of the development of HERG current blockade by telmisartan was investigated using an “envelope of tails” protocol[12,13]. Membrane potential was held at –80 mV and increased to +20 mV for increasing durations between 20 and 680 ms, after which HERG tails were evoked on repolarization to –40 mV. Original current traces before and after the addition of 50 µmol/L telmisartan are shown in Figure 3A and 3B. The fractional blockade of the tail currents, defined as the amplitude of tail currents reduced by the drug, divided by the control tail current amplitude at each duration site, was plotted with each time duration by a mono-exponential equation, and for HERG channels, it yielded a τ of 234.75±6.87 ms for the onset of post-depolarization channel blockade by 10 µmol/L (Figure 3C). We found that the levels of inhibition of the tail currents of HERG channels after blockade by 10 µmol/L telmisartan were enhanced from 0.27 at 40 ms and 0.30 at 400 ms to 0.37 at 680 ms. Our results suggested that the blockade was enhanced by further activation of currents, which were consistent with the open channel blockade.
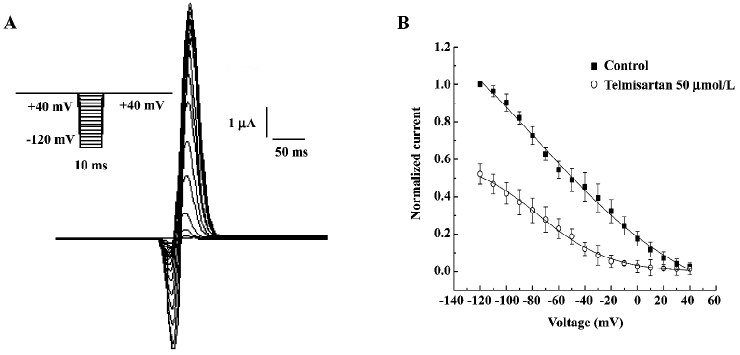
Effect of telmisartan on the inactivation of HERG channels in Xenopus oocytes The effect of telmisartan on the inactivation of HERG channels was assessed using a 3-step protocol shown in the inset of Figure 4A. After a 2 s pulse to +40 mV, the membrane potential was held to various test voltages from –120 mV to +40 mV for 10 ms to allow for the inactivation to relax to a steady state, and was then followed by a return step to +40 mV. The peak currents elicited by the second step to +40 mV provided a relative number of open conducting channels, while the re-inactivation of HERG was rapid at +40 mV pulse and was partially obscured by the capacitive currents (Figure 4A). Therefore, the peak outward currents during the third pulse was estimated by fitting current traces beginning 3 ms after depolarization with a mono-exponential equation, and extrapolating to the beginning of the pulse. Peak current amplitudes were then normalized to currents at –120 mV in the absence of 50 µmol/L telmisartan and then plotted as a function of the voltage of the 10 ms test pulse (steady-state inactivation curve) and fitted to a Boltzmann function. Peak current amplitudes of HERG channels were prominently decreased by 50 µmol/L telmisartan (Figure 4B,4C). The V1/2 (–76.63±13.56 mV vs –75.78±5.55 mV, n=6, P>0.05) for the steady-state inactivation was not significantly altered by telmisartan, and slope factors had a negative shift (62.34±12.82 mV vs 27.62±4.00 mV, n=6, P<0.05).
Effect of telmisartan on HERG channel deactivation kinetics in Xenopus oocytes Outward tail currents were recorded during a repolarization pulse from –120 mV to +10 mV, following a depolarization step to 0 mV in the absence or presence of 50 µmol/L telmisartan, respectively (Figure 5A). The time course of the deactivation tail currents was fitted with a double exponential function. The slow component of the deactivating tail currents was significantly lower in the presence of 10 and 50 µmol/L telmisartan in a concentration-dependent manner (n=6, P<0.05), while the fast component had no significant difference in the tested oocytes (n=6, P>0.05; Figure 5B, 5C). This indicated that telmisartan blocks the different component of HERG currents at different concentrations. The slow component of the deactivation in the presence of telmisartan suggested that closure of the activation gate was accelerated when the drug bound to the channels. This is different from the so-called “foot-in-the-door” effect[14], which occurs in many open-state channel blockers that block HERG channels. This gives us a better understanding that some HERG channel blockers may block HERG channels through a mechanism different from open-state blockade, which is similar to close-state blocking agent BeKm-1[15].
Discussion
In the present study, we analyzed the effects of telmisartan, a specific AT1 receptor antagonist, on Kv1.5 and HERG, which is involved in human cardiac repolarization. The maximum plasma concentrations obtained after administration of therapeutic doses of telmisartan (20–120 mg/d) was 0.04 to 1.15 µmol/L[2]. Thus this study demonstrated that at plasma concentration levels of therapeutic doses, telmisartan blocked hKv1.5 channels, whereas its effects on HERG channels occurred only at supra plasma concentration levels of therapeutic doses. It is important to note that the present experiments were performed in the absence of angiotensin. Moreover, even when the concentrations tested for each drug can be considered equipotent for AT1 receptor antagonism, the effects of telmisartan on each current differ in potency and in voltage and time dependency, which is not consistent with a common mechanism of action.
Effects of telmisartan on hKv1.5 channels Telmisartan exhibited a high affinity for hKv1.5 channels. However, the efficacy of blockade was different at peak and 1.5 s end-pulse currents, which were 7.75%±2.39% and 52.64%±3.77% at 1 µmol/L telmisartan, respectively. Furthermore, the concentration-dependent effects of telmisartan on hKv1.5 channels were biphasic, which was either the reduction in charge of crossing the membrane at positive potentials (open-channel interaction) or the inhibition of the currents elicited after conditioning pulses (interaction with the inactivated state). The reasons for this behavior are unknown, and experiments on site-directed hKv1.5 mutant channels would be needed to elucidate this issue. However, what cannot be excluded is that when the concentration of bulky molecules of telmisartan near the binding site increases, the steric hindrance interactions between them might decrease the efficacy of blockade. The importance of steric hindrance interactions in determining the blockade of quinidine at Kv1.4 channels has been demonstrated previously[16]. Telmisartan induced a voltage-dependent blockade on hKv1.5 channels that increased at the voltage range of channel activation. Moreover, telmisartan-induced blockade occurred in the range of potentials of channel opening. All these effects suggested an open-channel interaction. Furthermore, the blockade was significantly augmented with channel inactivation, suggesting that telmisartan also binds to the inactivated state. Affinity for both the open and the inactivation state has been previously described for irbesartan on Kv4.3 channels[17].
Effects of telmisartan on HERG channels Telmisartan blocked HERG channels in a concentration-dependent manner, and the IC50 value was 24.35 µmol/L at room temperature, which was approximately 21-fold higher than the free plasma concentration[2]. This result indicated that telmisartan had low affinity binding to HERG channels. This low affinity of telmisartan could be attributed to the spatial orientation of the most hydrophobic portion of the molecule (the spirocyclopentane ring), which was oriented perpendicularly to the imidazolone ring. Therefore, the telmisartan moiety would be rigid, preventing the proper interaction of the drug with its receptor site.
Our experiment with HERG channels suggested that telmisartan preferentially blocked open HERG channels, exhibiting several features that were typical of other open-channel blockers, such as dofetilide, chloroquine, and ziprasidone[14,18–20]. First, there was no blockade of initial step currents in response to depolarizing test pulses of –40 mV or more negative potentials. Meanwhile, in the different depolarization durations, the fractional blockade of peak tail currents at 40 ms duration was significantly lower than that at 400 and 680 ms, which indicated that following the channel activation, fractional blockade was enhanced by the activated channels. Second, the extent and rate of the onset of blockade was voltage dependent, increasing at more positive potentials even in the condition of leftward shift of the activation curve. These results also suggested that channel activation is required for telmisartan blockade.
Our research also demonstrated that the velocity of channel deactivation was significantly accelerated by telmisartan, which was inconsistent with the so-called “foot-in-the-door” effect. This indicated that telmisartan blocked HERG channels through a mechanism different from open-state blockade. This acceleration of the deactivation time course also occurred in BeKm-1 scorpion toxin, a close-state blocking agent of HERG channels. Therefore, telmisartan can also preferentially block close-state HERG channels.
Clinical significance HERG encodes the pore-forming subunits of channels that conduct rapid delayed rectifier K+ current IKr which is one of the most important membrane currents responsible for ventricular potential repolarization. The mutation of HERG causes long QT syndrome, a disorder of cardiomyocyte repolarization that predisposes affected individuals to an increased risk of torsades de pointes and lethal ventricular fibrillation[8,13,21,22]. Acquired long QT syndrome is far more common than inherited long QT syndrome and is often caused by the blockade of HERG channels treated with anti-arrhythmic agents and certain non-cardiac medications with diverse chemical structures and pharmacological actions[23,24]. It is widely believed that most drugs associated with torsades de pointes in humans are also associated with HERG K+ channel blockade at concentrations close to or superimposed upon the free plasma concentrations found in clinical use[25]. Our results demonstrate that the blockade of telmisartan on HERG channel occurred only at supra plasma concentration levels of therapeutic doses, which indicates that telmisartan may induce more little side-effects than other ARB, such as losartan, at therapeutic concentrations[26]. Telmisartan can also block hKv1.5 at plasma concentration levels of therapeutic doses. Kv1.5 channels are highly expressed in human atria (but not ventricles) and conduct ultrarapid delayed rectifier currents (Ikur) that contribute to action potential repolarization of human atrial myocytes[7,27,28]. Thus Kv1.5 is an important molecular target for the treatment of atrial fibrillation or atrial flutter, particularly because the inhibition of Kv1.5 should selectively prolong atrial but not ventricular action potential duration. Due to the dual effects, telmisartan may have good clinical security. However, as described for other ARB, multiple outward K+ currents participate in cardiac repolarization, and different ARB induce complicated effects simultaneously or successively. Further studies are needed to confirm their molecule mechanism, a significant undertaking that goes beyond the scope of the present manuscript.
Acknowledgements
We thank Michael C SANGUINETTI for providing the cDNA of HERG channel, Dr Maria L GARCIA for the hKv1.5, and He-ping GUO for technical and equipment support.
References
- Burnier M, Brunner HR. Angiotensin II receptor antagonists. Lancet 2000;355:637-45.
- Stangier J, Su C, Roth W. Pharmacokinetics of orally and intravenously administered telmisartan in healthy young and elderly volunteers and in hypertensive patients. J Int Med Res 2000;28:149-67.
- Delpón E, Caballero R, Gómez R, Núñez L, Tamargo J. Angiotensin II, angiotensin II antagonists and spironolactone and their modulation of cardiac repolarization. Trends Pharmacol Sci 2005;26:155-61.
- Tristani-Firouzi M, Sanguinetti MC. Structural determinants and biophysical properties of HERG and KCNQ1 channel gating. J Mol Cell Cardiol 2003;35:27-35.
- Roden DM, George AL Jr. Structure and function of cardiac sodium and potassium channels. Am J Physiol 1997;273:H511-25.
- Tamkun MM, Knoth KM, Walbridge JA, Kroemer H, Roden DM, Glover DM. Molecular cloning and characterization of two voltage-gated K+ channel cDNAs from human ventricle. FASEB J 1991;5:331-7.
- Wang Z, Fermini B, Nattel S. Sustained depolarization-induced outward current in human atrial myocytes. Evidence for a novel delayed rectifier K+ current similar to Kv1.5 cloned channel currents. Circ Res 1993;73:1061-76.
- Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 1995;81:299-307.
- Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW, et al. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell 1999;97:175-87.
- Caballero R, Delpón E, Valenzuela C, Longobardo M, Tamargo J. Losartan and its metabolite E3174 modify cardiac delayed rectifier K+ currents. Circulation 2000;101:1199-205.
- Wang X, Liao Y, Zou A, Li L, Tu D. Blockade action of ketanserin and increasing effect of potassium ion on Kv1.3 channels expressed in Xenopus oocytes. Pharmacol Res 2007;56:148-54.
- Milnes JT, Crociani O, Arcangeli A, Hancox JC, Witchel HJ. Blockade of HERG potassium currents by fluvoxamine: incomplete attenuation by S6 mutations at F656 or Y652. Br J Pharmacol 2003;139:887-98.
- Trudeau MC, Warmke JW, Ganetzky B, Robertson GA. HERG, a human inward rectifier in the voltage-gated potassium channel family. Science 1995;269:92-5.
- Sanchez-Chapula JA, Navarro-Polanco RA, Culberson C, Chen J, Sanguinetti MC. Molecular determinants of voltage-dependent human ether-a-go-go related gene (HERG) K channel block. J Biol Chem 2002; 277: 23 587–95.
- Milnes JT, Dempsey CE, Ridley JM, Crociani O, Arcangeli A, Hancox JC, et al. Preferential closed channel blockade of HERG potassium currents by chemically synthesised BeKm-1 scorpion toxin. FEBS Lett 2003;547:20-6.
- Zhang H, Zhu B, Yao JA, Tseng GN. Differential effects of S6 mutations on binding of quinidine and 4-aminopyridine to rat isoform of Kv1.4: common site but different factors in determining blockers’ binding affinity. J Pharmacol Exp Ther 1998;287:332-43.
- Moreno I, Caballero R, González T, Arias C, Valenzuela C, Iriepa I, et al. Effects of irbesartan on cloned potassium channels involved in human cardiac repolarization. J Pharmacol Exp Ther 2003;304:862-73.
- Mitcheson JS, Chen J, Lin M, Culberson C, Sanguinetti MC. A structural basis for drug-induced long QT syndrome. Proc Natl Acad Sci USA 2000;97:12329-33.
- Su Z, Chen J, Martin RL, McDermott JS, Cox BF, Gopalakrishnan M, et al. Block of hERG channel by ziprasidone: biophysical properties and molecular determinants. Biochem Pharmacol 2006;71:278-86.
- Sanchez-Chapula JA, Ferrer T, Navarro-Polanco RA, Sanguinetti MC. Voltage-dependent profile of human ether-a-go-go-related gene channel block is influenced by a single residue in the S6 transmembrane domain. Mol Pharmacol 2003;63:1051-8.
- Keating MT, Sanguinetti MC. Molecular and cellular mechanisms of cardiac arrhythmias. Cell 2001;104:569-80.
- Iskin S. Long QT syndromes and torsade de pointes. Lancet 1999;354:1625-33.
- Cavero I, Mestre M, Guillon JM, Crumb W. Drugs that prolong QT interval as an unwanted effect: assessing their likelihood of inducing hazardous cardiac dysrhythmias. Expert Opin Pharmacother 2000;1:947-73.
- De Ponti F, Poluzzi E, Cavalli A, Recanatini M, Montanaro N. Safety of non-antiarrhythmic drugs that prolong the QT interval or induce torsade de pointes: an overview. Drug Saf 2002;25:263-86.
- Redfern WS, Carlsson L, Davis AS, Lynch WG, MacKenzie I, Palethorpe S, et al. Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovasc Res 2003;58:32-45.
- Caballero R, Delpón E, Valenzuela C, Longobardo M, Tamargo J. Losartan and its metabolite E3174 modify cardiac delayed rectifier K+ currents. Circulation 2000;101:1199-205.
- Fedida D, Wible B, Wang Z, Fermini B, Faust F, Nattel S, et al. Identity of a novel delayed rectifier current from human heart with a cloned K+ channel current. Circ Res 1993;73:210-6.
- Snyders DJ, Tamkun MM, Bennett PB. A rapidly activating and slowly inactivating potassium channel cloned from human heart. Functional analysis after stable mammalian cell culture expression. J Gen Physiol 1993;101:513-43.