
Nitrofen suppresses cell proliferation and promotes mitochondria-mediated apoptosis in type II pneumocytes1
Introduction
Nitrofen (2, 4-dichloro-40-nitrodiphenyl ether) is a selective contact herbicide used on a variety of food crops for pre- and post-emergence control of annual grasses and weeds[1]. It is a member of the chlorophenoxy class of herbicides and is also called nitrophene, TOK, TOK E-25, and Nip[1]. Nitrofen is a potent teratogen in rats that produces abnormal development of the heart, kidneys, diaphragm, and lung when administered during organogenesis[2]. It has been demonstrated in pregnant rodents that nitrofen induces congenital diaphragmatic hernia (CDH)[3], including pulmonary hypoplasia[4] with abnormal pulmonary arterioles and biochemical lung immaturity[5] similar to those observed in human CDH. The reliable induction of CDH and pulmonary hypoplasia-immaturity in fetal rodents exposed to nitrofen has made this teratogenic model the main tool for understanding the pathogenesis of CDH.
Previous evidence indicates that the lung is malformed independently, and perhaps is the primary cause of diaphragmatic anomalies in CDH[6,7]. It has been clearly shown that nitrofen on its own can, to varying degrees, affect aspects of lung development[8]. Nitrofen levels in the embryo are likely persist for several days after administration, allowing multiple stages of lung embryogenesis to be targeted by this teratogen throughout the final third of rat gestation[8]. Recent data have suggested that nitrofen may interfere with retinoid signaling during lung development, as measured with transgenic mice containing the Lac Z reporter gene linked to a retinoic acid response element[9]. Inadequate retinoic acid signaling resulted in incomplete differentiation cells and ultimately cell death[9]. However, the mechanisms of nitrofen-mediated pulmonary hypoplasia are still incompletely understood.
In developing and adult lungs, type II pneumocytes are not only the source of alveolar surfactant, but also the progenitor cells for alveolar epithelium[10]. Type II pneumocytes produce and secrete pulmonary surfactant, which is critical for effective gas exchange[10]. Type II pneumocytes also proliferate and differentiate into type I pneumocytes to restore the alveolar epithelium after lung injury and participate in the innate immune response to exogenous materials and organisms[10]. Li et al demonstrated that in the lungs of nitrofen-induced CDH, type II pneumocytes were filled with only a few lamellar bodies and become metabolically inactive[11]. The sparse type II pneumocytes also showed cytoplasmic degenerative changes, such as vacuolization of the cytoplasm[11]. In cultured type II pneumocytes, nitrofen decreased synthesis of surfactant components like surfactant protein B, and downregulated thyroid transcription factor-1, a homeotic protein that acts as a transcription factor during pulmonary morphogenesis[12]. These findings show that nitrofen-induced pulmonary hypoplasia is exerted, at least in part, by direct action on type II pneumocytes[12]. However, it still remains largely unknown whether nitrofen exerts effects on proliferation and apoptosis of type II pneumocytes. In the present study, we demonstrated that nitrofen suppressed proliferation of cultured A549 cells accompanied by the downregulation of proliferating cell nuclear antigen (PCNA), and induced mitochondria-mediated apoptosis involving the activation of the p38 mitogen-activated protein kinase (p38-MAPK) signaling pathway.
Materials and methods
Cell culture and nitrofen treatment A549, a type II pneumocyte cell line with phenotypic features including surfactant protein synthesis[13], was obtained from ATCC (CCL-185, Manassas, VA, USA) and maintained in Dulbecco’s minimal Eagle’s medium (DMEM, Life Technologies Inc, Gaithersburg, MD, USA) supplemented with 10% fetal bovine serum (FBS, Life Technologies, USA), penicillin (100 U/mL), and streptomycin (100 µg/mL). The cells were maintained at 37 °C in a humidified atmosphere of 5% CO2. Confluent monolayers of cells were starved with a medium supplemented with 0.1% FBS and 2 mmol/L L-glutamine. Twenty-four hours later, the cells were incubated in serum-free DMEM for 4 h and pretreated with Z-Val-Ala-Asp(OCH3)-fluoromethylketone (zVAD-fmk, Biomol, Plymouth Meeting, PA, USA) or SB203580 (Calbiochem, La Jolla, CA, USA) for 1h, then stimulated with different concentrations of 2,4-dichloro-40-nitrodiphenyl ether (nitrofen, Sigma, St Louis, MO, USA) or DMSO as indicated.
Measurement of cell viability For direct cell counting, 1×104 A549 cells were seeded in 24-well plates, and treated with nitrofen or DMSO as indicated. At the end of the incubation period, the cells were washed with phosphate-buffered saline (PBS), trypsinized, and counted using a Casy 1-model TT cell counter (Schaefer System GmbH, Reutlingen, Germany). Cell proliferation was monitored by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT, Sigma, USA) colorimetric assay[14]. Briefly, 20 µL MTT (5 mg/mL) was added to each well. After 4 h incubation at 37 ºC, the cell supernatant was discarded, the MTT crystals were dissolved with DMSO, and the absorbance was measured at 570 nm. Percent viability was defined as the relative absorbance of treated versus untreated control cells. Cellular DNA synthesis was determined by [3H]-thymidine incorporation assay. Briefly, the cells (2×105/well) were grown on 24-well plates. A pulse of 1 µCi/mL [3H]-thymidine (DuPont, Boston, MA, USA; sp act 80–90 Ci/mmol) was added to the cells for 1 h. DNA synthesis was determined as trichloroacetic acid (TPA)-precipitable incorporation of [3H]-thymidine as described by Sheffield[15]. All experiments were done with 6–8 wells per experiment and repeated at least 3 times.
Colony formation assay The cells were seeded at a density of 500 mL on 35 mm dishes[16]. After an overnight incubation to allow cell attachment, the cells were incubated with nitrofen or DMSO as indicated. After being incubated for 24 h, the medium was replaced with fresh medium containing 10% FBS. The colonies were allowed to grow for 10–14 d. The medium was discarded and each well was washed twice with PBS carefully. The cells were fixed in methanol for 15 min and then stained with crystal violet for 20 min. Finally, positive colony formation (more than 50 cells/colony) was counted. The survival fraction for the cells was expressed as the ratio of plating efficiency of treated cells to that of untreated control cells.
Cell cycle assay According to the literature[17], cell cycles in untreated, DMSO- or nitrofen-treated cells were examined by flow cytometry. Briefly, 2×105 cells were collected, washed twice with PBS, and fixed in 70% ethanol overnight at 4 °C. Then the cells were washed once with PBS, digested with 200 µL RNase (1 mg/mL) at 37 °C for 30 min, and stained with 800 µL propidium iodide (PI, 50 µg/mL, Sigma, USA) at room temperature for 30 min. The DNA histograms were assayed with a flow cytometer (Becton-Dickinson, San Jose, CA, USA), using the CELLQUEST software (Becton-Dickinson, USA).
Cellular morphological observation To observe the changes in cellular morphology, the in situ terminal deoxynucleotidyl transferase-mediated dUTP nick-end-labeling (TUNEL, Roche, Indianapolis, IN, USA) method was performed according to the manufacturer’s instructions. Briefly, the cells were fixed immediately in 4% paraformaldehyde for 20 min, washed with PBS, and permeabilized with 0.1% Triton X-100 in 0.1% sodium citrate. Each of the sample slides received 50 µL TUNEL reaction mixture and was incubated for 60 min at 37 ºC. After washing with PBS, the sections were analyzed under a Leitz fluorescent microscope (Leitz, Wetzlar, Germany). Negative control slides were performed without the TUNEL mixture. The TUNEL positively-stained cells were counted in 10 randomly selected high-power (×200) fields. The rate of the positively stained cells was determined by calculating the average percentage.
Morphological evidence of apoptosis was further obtained using acridine orange (AO, Sigma, USA) and ethidium bromide (EB, Sigma, USA) staining[18]. Briefly, the cells were harvested with 0.125% trypsin and 0.01% EDTA, resuspended in 95 µL DMEM medium, and incubated with 5 µL AO/EB staining solution (100 mg/L PBS of each dye) at room temperature for 15 min. The cells were examined using fluorescence microscopy and photographed (Olympus, Tokyo, Japan). Viable cells were colored green with intact nuclei. Nonviable cells had bright orange chromatin. Apoptosis was demonstrated by the appearance of cell shrinkage with condensation and fragmentation of the nuclei. Necrotic cells appeared orange with a normal nuclear structure. The numbers of viable cells with normal nuclei (VN), viable cells with apoptotic nuclei (VA), nonviable cells with apoptotic nuclei(NVA), and nonviable cells with normal nuclei cells (NVN) were determined by counting 5 randomly selected high-power (×200) fields. Apoptosis rates were calculated as (%)=(VA+ NVA)/(VN+ NVN+VA+NVA)×100%.
Apoptosis rate detection The apoptotic ratios of cells were determined by annexin V- fluorescein isothiocyanate (FITC) (BD Pharmingen, San Diego, CA, USA) and PI staining flow cytometry[18]. Briefly, the cells from the above groups were collected, washed twice with cold PBS, resuspended with 100 µL binding buffer [10 mmol/L 4-(2-Hydroxy-ethyl)-1-piperazineethanesulfonic acid, 140 mmol/L NaCl, and 2.5 mmol/L CaCl2, pH 7.4] into 2×105–5×105 cells/mL density, and incubated with annexin V-FITC at room temperature for 10 min. After washing with the binding buffer, the cells were resuspended in 400 µL binding buffer containing 10 µL PI (20 µg/mL, Sigma, USA), and incubated on ice for 15 min. Apoptosis was analyzed by flow cytometry (Becton-Dickinson, USA) at a wavelength of 488 nm. This method can be used to distinguish between living cells (annexin V–/PI–), early apoptotic/primary apoptotic cells (annexin V+/PI–), late apoptotic/secondary necrotic cells (annexin V+/PI+), and necrotic cells (annexin V–/PI+)[19].
Measurement of mitochondrial membrane potential (Δψ m) The mitochondrial membrane potential was analyzed using 5,5',6,6'- tetrachloro-1,1',3,3'-tetraethylbenzimidazolyl- carbocyanine iodide (JC-1, Sigma, USA), a lipophilic cationic fluorescence dye. JC-1 exists as a green fluorescent monomer (emission wavelength is 527 nm) at low mitochondrial membrane potential. Mitochondrial depolarization is indicated by an increase in green fluorescence (FL-1)[20]. The cells (1×106) were incubated with 5 mg/mL JC-1 for 15 min at room temperature in dark conditions. After centrifugation at 200×g for 5 min, the cells were washed twice with PBS at 4 °C, resuspended in 0.5 mL PBS, and analyzed on a flow cytometer (Becton-Dickinson, USA).
Immunofluorescence for apoptosis-inducing factor (AIF) The cells were grown on sterile coverslips in a 6-well plate and treated with nitrofen as indicated. The cells were fixed with fresh 4% formaldehyde in PBS at 4 °C for 20 min and permeabilized with pre-chilled PBS with 0.2% Triton X-100. The cells were then incubated with a rabbit antibody against AIF (1:100, Santa Cruz Biotechnology, Santa Cruz, CA, USA) in 3% bovine serum albumin/PBS overnight at 4 °C followed by incubation with fluorescein isothiocyanate-conjugated secondary antibodies. After PBS washes, nuclei were counterstained with PI. Images were captured on a fluorescent microscope.
Real-time RT-PCR for PCNA Total RNA was isolated with a RNeasy mini kit (Qiagen, Valencia, CA, USA). The RT reactions were conducted with a Transcriptor First Strand cDNA Synthesis kit (Roche, USA). Real-time PCR with SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA) was performed using the ABI Prism 7700 Sequence Detector (Applied Biosystems, USA). The PCR primers for PCNA were as follows: 5'-AAACTAGCTAGACTTTCCTC-3' and 5'-TCACGCCCATGGCC AGGTTG-3', amplifying a 274 bp fragment.
Western blotting for PCNA, caspase-3, Bcl-2, Bcl-xL, Bax, Bak, AIF, and phosphorylation of the p38 kinase The cells were collected and extracted with 1×cell lysis buffer (Promega, Madison, WI, USA). Nuclei and mitochondria were isolated according to reported protocols[21]. Protein (50 µg) from each sample was subjected to 4%–20% pre-cast polyacrylamide gel (Bio-Rad, Hercules, CA, USA) electrophoresis and transferred to nitrocellulose membranes (Bio-Rad, USA). For PCNA, caspase-3, Bcl-2, Bcl-xL, Bax, Bak, and AIF (Santa Cruz Biotechnology, USA) detections, the specific primary antibody dilution was 1:1000, 1:500, 1:500, 1:500, 1:500, 1:500, and 1:500, respectively. For the phosphorylation assay of the p38 kinase, the membranes were probed with the specific primary antibodies against the phospho-specific and non-phosphorylated p38 kinase at a dilution of 1:500 (Santa Cruz Biotechnology, USA). The second antibody used in this assay was the goat anti-rabbit horseradish peroxidase -labeled antibody at a dilution of 1:3000 (Bio-Rad, USA). An enhanced chemiluminescence substrate kit (Amersham, Piscataway, NJ, USA) was used for the chemiluminescent detection of the signals with autoradiography film (Amersham, USA).
Statistical analysis Unless otherwise stated, all data were shown as mean±SEM. Statistical significance (P<0.05) was determined by t-test or ANOVA followed by an assessment of differences using SigmaStat 2.03 software (Jandel, Erkrath, Germany).
Results
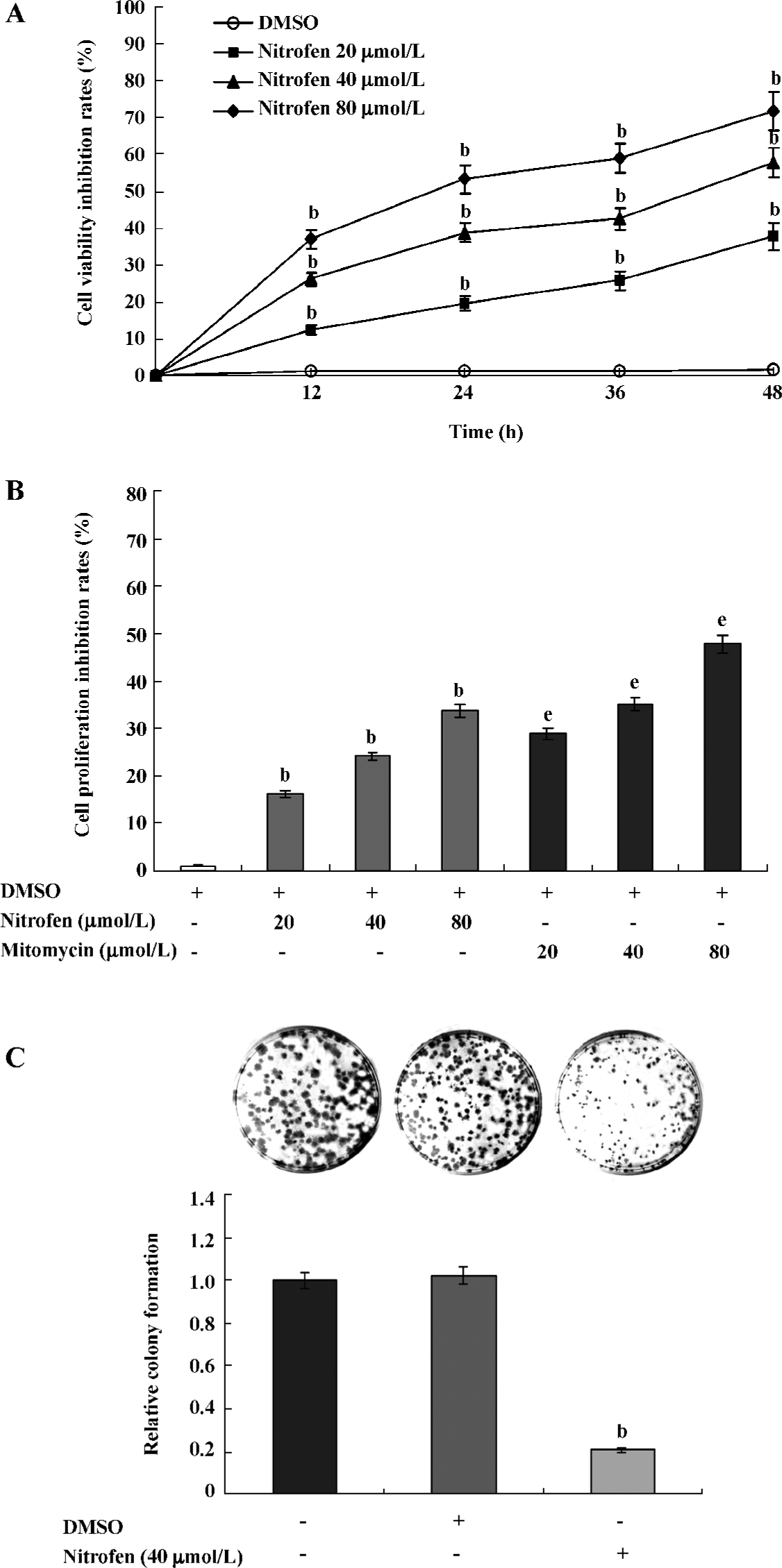
Nitrofen suppressed the proliferation of A549 cells Precisely controlled cell proliferation and apoptosis are prerequisites for normal development and homeostasis of lungs. Abnormalities in these processes are emerging to explain features of disorders, including pulmonary hypoplasia[22]. To explore the effects of nitrofen on type II pneumocytes, we first observed cell proliferation changes in the cultured A549 cell line. As shown in Figure 1A, nitrofen administration resulted in distinct decreases of cell viability in a dose- and time-dependent manner. However, DMSO treatment led to no significant change in cell viability of A549 cells. MTT colorimetric assay indicated that nitrofen inhibited the proliferation of A549 cells, which was lower than those induced by mitomycin as the positive controls (Figure 1B). The colony formation assay further demonstrated the cell proliferation inhibition effects of nitrofen in cultured A549 pneumocytes (Figure 1C). These findings indicated that nitrofen suppressed the in vitro cell proliferation of cultured type II pneumocytes.
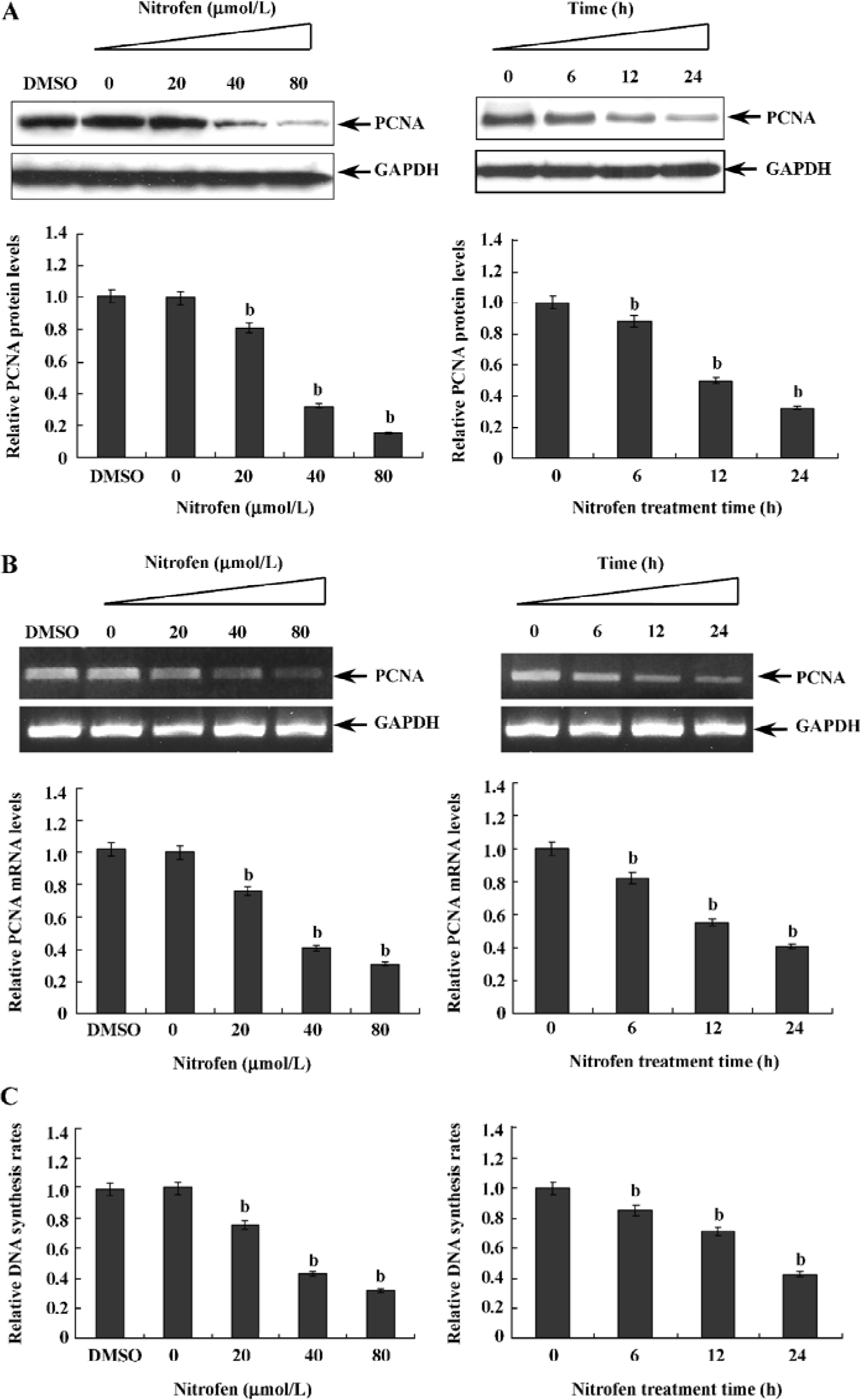
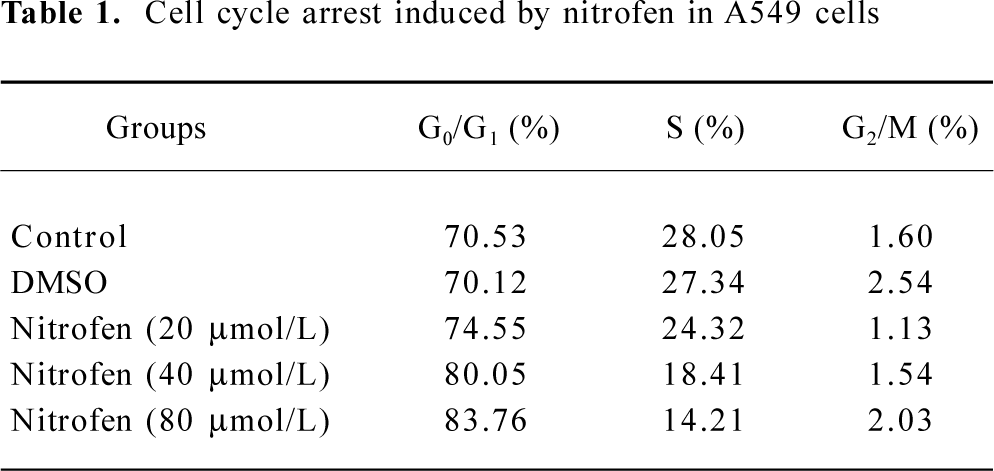
Downregulation of PCNA might be associated with nitrofen-induced decreases in the proliferation of A549 cells PCNA is an intranuclear polypeptide maximally synthesized during the S-phase of cell cycles and participates in cell proliferation[23]. Since the above evidence indicated that nitrofen suppressed cell proliferation of type II pneumocytes, we hypothesized that nitrofen may modulate PCNA expression in these cells. To meet this end, A549 cells were treated with various concentrations of nitrofen for different periods. As shown in Figure 2A, Western blotting indicated that nitrofen downregulated the expression level of PCNA in A549 cells in a dose- and time-dependent manner. However, DMSO administration had no influence on the expression of PCNA (Figure 2A). In addition, the results of real-time RT-PCR indicated that nitrofen can induce decreases of PCNA mRNA in A549 cells (Figure 2B). Consistent with these findings, the DNA synthesis of nitrofen-treated A549 cells was attenuated (Figure 2C). Moreover, nitrofen treatment reduced the ratios of the S-phase, and induced G0/G1 arrest in cultured A549 cells (Table 1). These findings indicated that nitrofen-induced decreases in cell proliferation might be associated, at least in part, with the downregulation of PCNA.
Full table
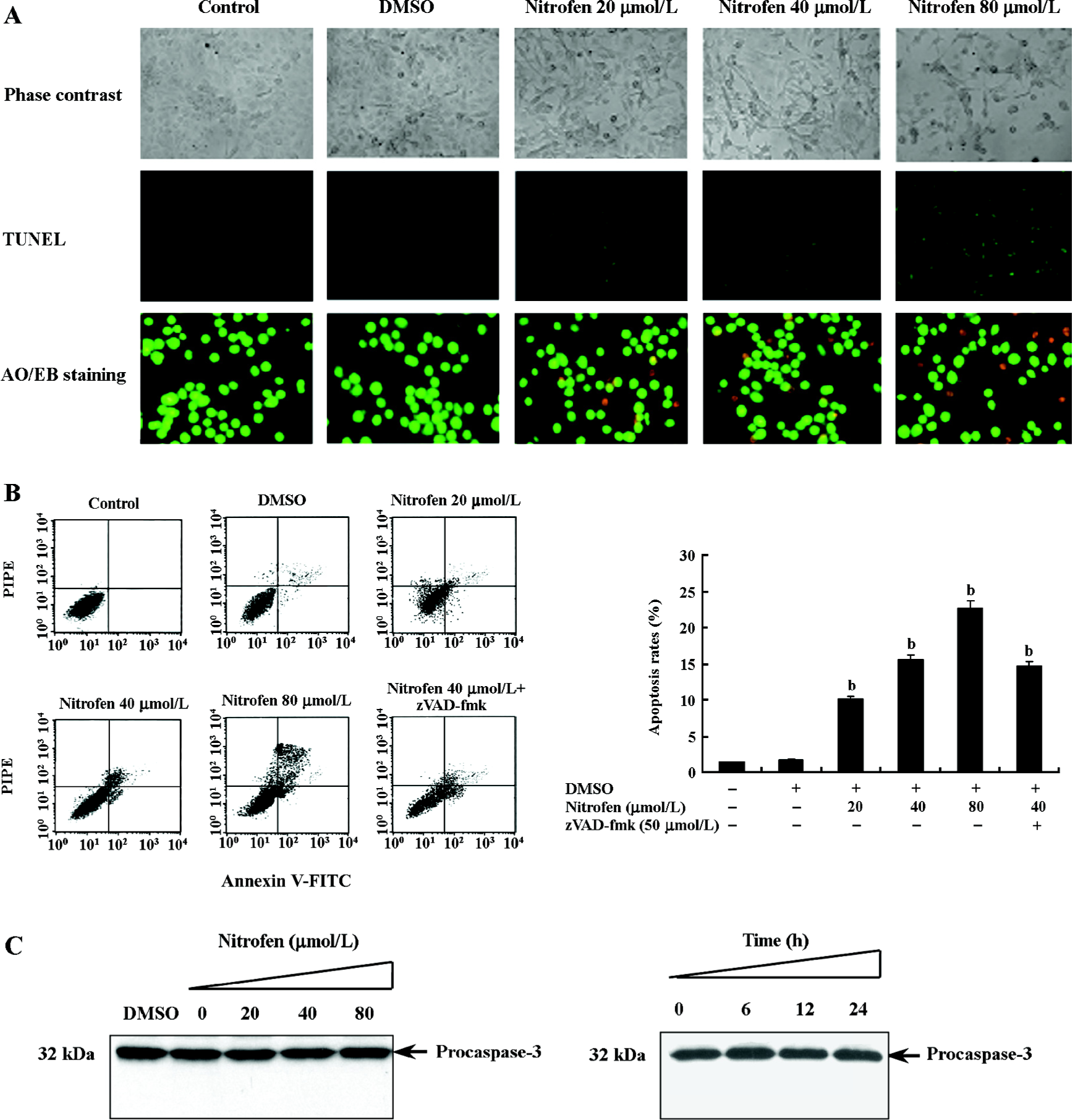
Nitrofen induced apoptosis in A549 cells in a caspase-independent manner Previous studies demonstrated enhanced apoptosis in the cervical somites of nitrofen-exposed rat embryos as a mechanism for diaphragmatic maldevelopment in CDH[24]. We hypothesized in this study that disturbed apoptosis may participate in nitrofen-induced cell death in cultured type II pneumocytes. As shown in Figure 3A, TUNEL indicated that treatment of A549 cells with nitrofen resulted in an obvious DNA strand break, a characteristic change of apoptosis. To further investigate the type of cell death induced by nitrofen treatment, the cells were stained with AO/EB, which allows the identification of viable, apoptotic, and necrotic cells based on color and appearance[18]. As shown in Figure 3A, the treatment of A549 cells with nitrofen for 24 h resulted in nuclei-shrunk and orange-stained cells. However, after treatment with DMSO for 24 h, there were no such morphological changes in the A549 cells (Figure 3A). This procedure and annexin V-FITC/PI staining flow cytometry were also used to quantify the number of apoptotic cells induced by nitrofen treatment. As shown in Table 2 and Figure 3B, 20–80 µmol/L nitrofen exerted strong apoptosis-inducing effects on A549 cells. Additionally, pretreatment of A549 cells with zVAD-fmk (50 µmol/L), a pan-caspase inhibitor[25], did not abolish nitrofen-induced apoptosis (Table 2; Figure 3B). Furthermore, Western blotting indicated that nitrofen administration would not influence the expression level and cleavage of procaspase-3 within A549 cells (Figure 3C). These findings indicated that a caspase-independent mechanism was involved in nitrofen-induced apoptosis.
Full table
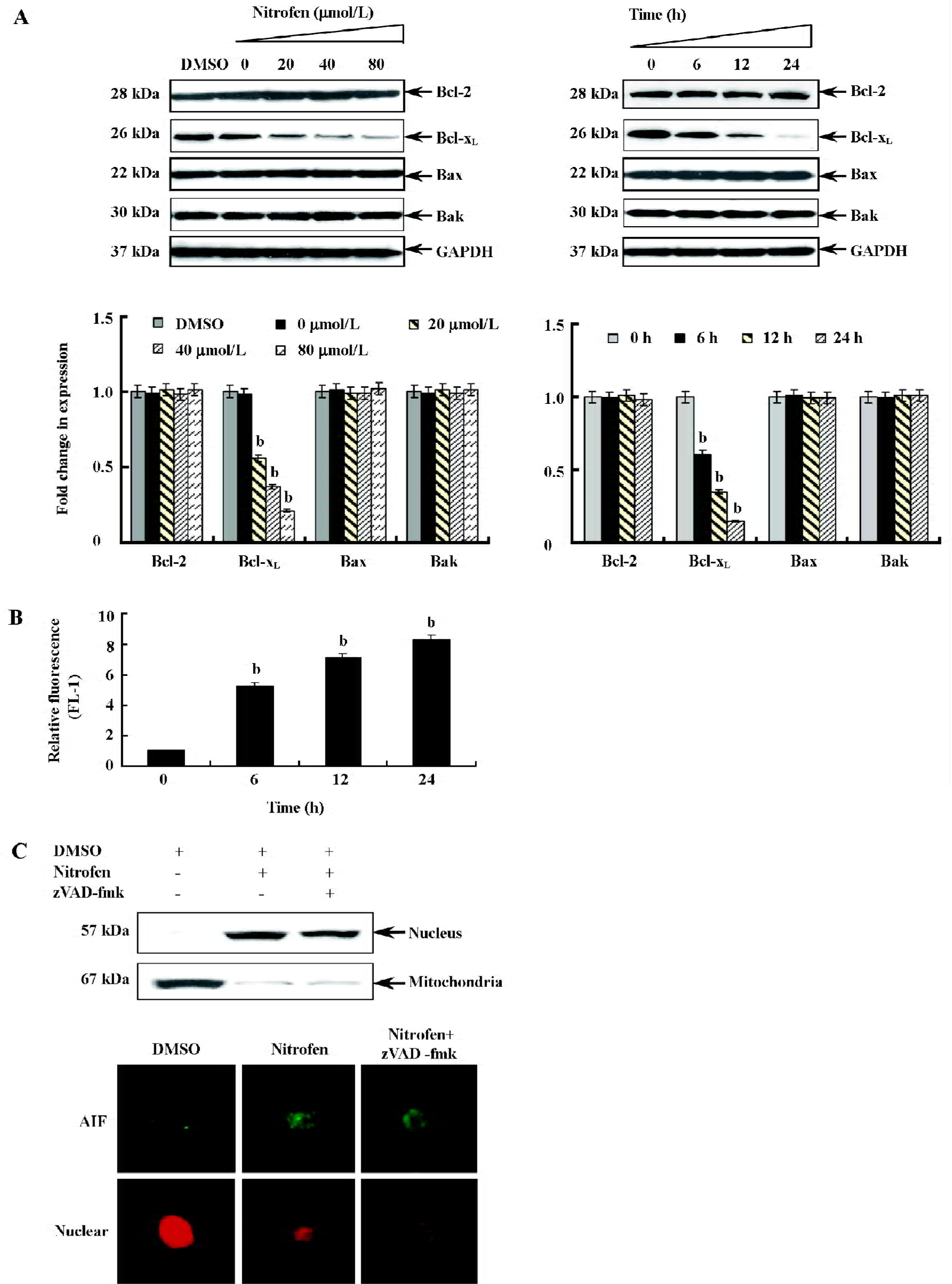
Participation of the mitochondria-mediated pathway in nitrofen-induced apoptosis in A549 cells To determine the involvement of the mitochondria-mediated pathway in nitrofen-induced apoptotic cell death, we first performed Western blotting to measure the expression of Bcl-2, Bcl-xL, Bax, and Bak, which are members of the Bcl-2 family proteins critical to maintaining the integrity of the mitochondrial membrane (Figure 4A). There was a decrease in the expression of anti-apoptotic Bcl-xL in a dose- and time-dependent manner (Figure 4A). The decrease was significant after 6 h, and at 24 h, expression decreased to 15.2% compared to the control (Figure 4A). However, there was no alteration in the expression of Bcl-2, Bax, and Bak (Figure 4A). Then we measured changes in mitochondrial membrane potential (Δψm). As shown in Figure 4B, nitrofen treatment of A549 cells resulted in a rapid dissipation of Δψm in a time-dependent manner with an increase in green fluorescence emission. Previous studies demonstrated that a loss of permeability of the mitochondrial membrane leads to the release of apoptogenic proteins normally confined to mitochondrial intermembrane space[21]. AIF is synthesized as a 67 kDa preprotein and localizes in mitochondrial intermembrane space[26]. Upon induction of apoptosis, AIF is processed to a 57 kDa form, translocated into the nucleus, and plays important roles in caspase-independent apoptosis[26]. We hypothesize that AIF may participate in nitrofen-induced apoptosis. Western blotting and immunofluorescence studies confirmed that nitrofen administration lead to the translocation of AIF from the mitochondria to the nucleus in A549 cells (Figure 4C). Moreover, pretreatment with the general caspase inhibitor zVAD-fmk (50 µmol/L) did not prevent the AIF translocation (Figure 4C). These results indicated that the mitochondria-mediated pathway participated in nitrofen-induced apoptosis.
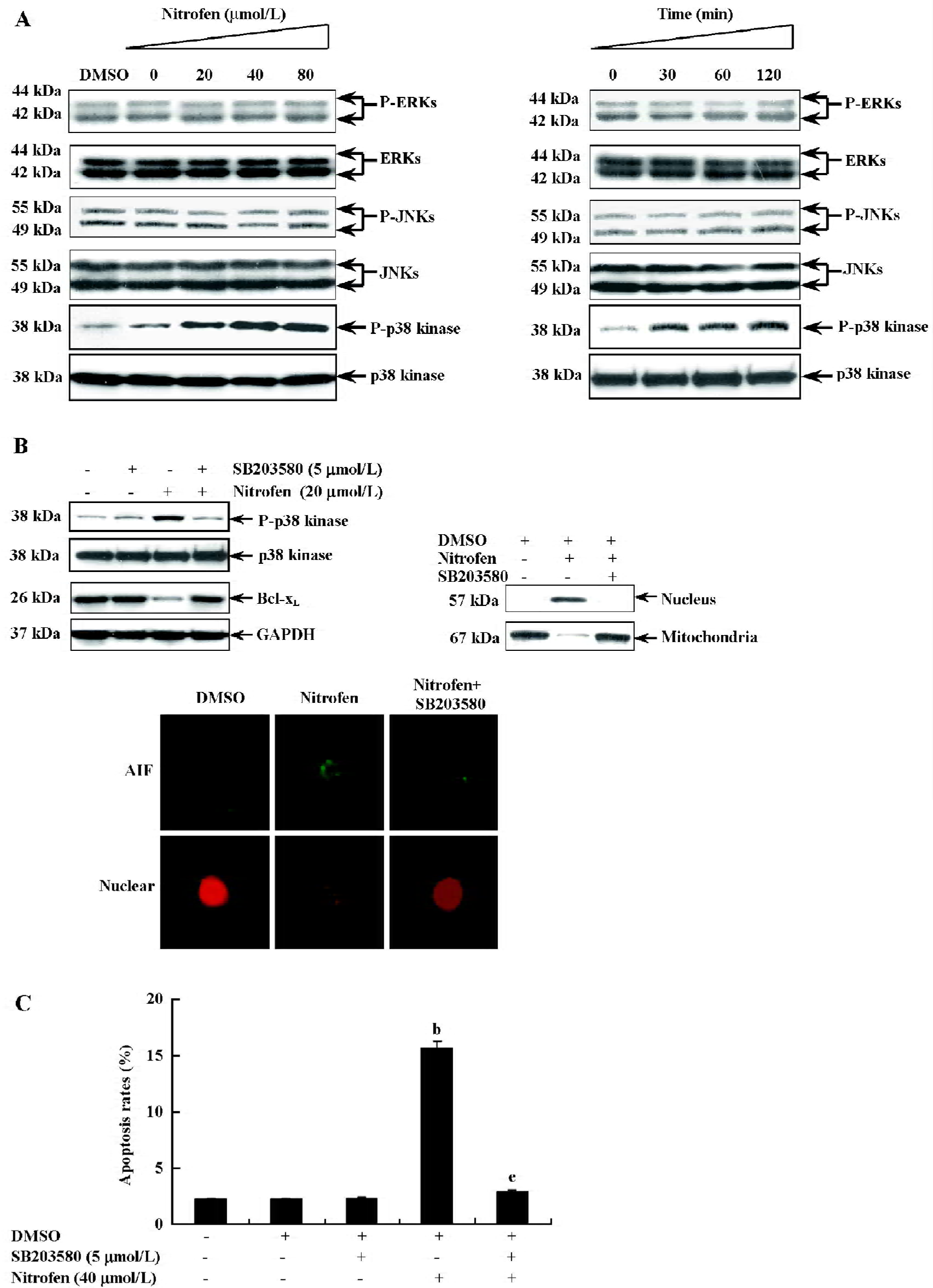
Involvement of the p38-MAPK signaling pathway in nitrofen-induced apoptosis in A549 cells Apoptosis has been demonstrated to be associated with changes in MAPK activity in a number of different cell systems[27,28]. To determine whether MAPK were involved in nitrofen-induced apoptosis, its activation was evaluated by Western blot analysis with antibodies specific for the phosphorylated (activated) forms of MAPK. As shown in Figure 5A, nitrofen strongly induced the phosphorylation of p38-MAPK. The activation of extracellular signal-regulated protein kinase (ERK)1/2 and the c-Jun N-terminal kinase (JNK) were examined using the same extracts, but did not show changes in phosphorylation, suggesting that ERK-1/2 and JNK were not affected by nitrofen (Figure 5A). To clarify whether mitochondria-mediated apoptosis induced by nitrofen is dependent upon p38-MAPK activation, the p38-MAPK inhibitor SB203580 was used to treat A549 cells prior to nitrofen exposure. As shown in Figure 5B, the incubation of cells with SB203580 (5 µmmol/L) blocked the nitrofen-induced phosphorylation of p38-MAPK and downregulation of Bcl-xL, resulting in rescued nuclear translocation of AIF. In addition, pretreatment of A549 cells with SB203580 (5 µmol/L) abolished nitrofen-induced apoptosis (Figure 5C). These findings indicate that activated p38-MAPK is involved in nitrofen-induced apoptosis in A549 cells.
Discussion
PCNA is a protein that acts in conjunction with DNA polymerase delta during mitosis[29]. It also plays a central role in DNA replication, DNA repair, and cell cycle progression[29]. Until now, PCNA expression has been an established operational marker for proliferating cells. In the current study, we demonstrated that nitrofen treatment suppressed the cell proliferation of cultured pneumocytes by cell counting, MTT colorimetry, and colony formation assay (Figure 1). In addition, we found that nitrofen downregulated the transcription and expression level of PCNA in cultured pneumocytes in a dose- and time-dependent manner (Figure 2A,B), which was consistent with the findings of Keijzer et al that when lung explants were treated with nitrofen, PCNA immunoreactivity was clearly reduced in the mesenchymal component[30]. In addition, PCNA reactivity was only observed perinuclear and in the cytoplasm of epithelial cells of the explants exposed to nitrofen[30]. In the present study, we also demonstrated that nitrofen treatment could induce G0/G1 arrest and reduce S-phase cells by flow cytometry (Table 1), which was consistent with previous findings that PCNA has several roles in progression through the S phase[29]. Moreover, DNA synthesis of nitrofen-treated A549 cells decreased compared to those of the controls (Figure 2C). These results indicated that proliferation is disturbed in cultured type II pneumocytes exposed to nitrofen, which might be associated with the downregulation of PCNA. Impaired regulation of cell proliferation may therefore underpin nitrofen-induced pulmonary hypoplasia in CDH.
Apoptosis is a controlled type of cell death characterized by cell shrinkage, membrane blebbing, and DNA fragmentation. Apoptosis is the result of coordinated signaling pathways, which can be triggered by a variety of extracellular stimuli, such as radiation, cytokines, and growth factor withdrawal[31]. We hypothesized in this study that the disturbed apoptosis may participate in nitrofen-induced pulmonary hypoplasia. Although a previous study examined the possibility of nitrofen-mediated apoptosis in type II H441 pneumocytes, it found no observable increases in apoptosis by TUNEL assay[12]. Our present study found that nitrofen, with high concentrations (20–80 µmol/L) rather than low concentrations (1.5 µmol/L), can induce apoptosis of A549 cells as demonstrated not only by TUNEL, but also by AO/EB assay and flow cytometry (Figure 3A,B; Table 2). In a recent report, Kling et al demonstrated that administration of nitrofen to teratocarcinoma P19 cells resulted in caspase-3 cleavage and caspase-dependent apoptosis[32]. However, in the present study we observed no cleavage of caspase-3, the key executor of apoptotic cell death, within nitrofen-treated type II A549 pneumocytes (Figure 3C). In addition, pretreatment of A549 cells with zVAD-fmk, a pan-caspase inhibitor[25], did not abolish nitrofen-induced apoptosis (Figure 3B), which indicated that a caspase-independent mechanism is involved in nitrofen-induced apoptosis of type II pneumocytes. We analyzed that the different findings between the current study and Kling’s report[32] may be due to the characteristic of different cell types. Recent evidence indicates that differentiated cells appear to be more resistant to caspase-dependent apoptosis than the undifferentiated ones[33]. Teratocarcinoma cells, including the P19 cell line, are pluripotent and exhibit similar characteristics to undifferentiated embryonic and fetal cell types[34]. However, the A549 pneumocytes are likely more differentiated[13] and consequently, might be resistant to nitrofen-induced caspase machinery, in which case the caspase-independent pathway is triggered and enrolled in apoptosis.
In recent years, mitochondria are thought to act as key coordinators of cell death. Numerous studies have demonstrated that most cells manifest a collapse of mitochondrial membrane potential as a prelude to nuclear DNA degradation and apoptosis[35]. Several pro-apoptotic signaling and damage pathways converge on mitochondria to induce mitochondrial membrane permeabilization, and the responsible molecules are Bcl-2 family proteins[36]. In healthy cells, Bax and Bak are in an inactive conformation in which their NH2 and COOH termini are folded into a hydrophobic pocket[34]. In response to apoptotic stimuli, these proteins unfold and form multimers in the mitochondrial membrane. The lethality of this multimerization is blocked through the formation of heterodimers with Bcl-2 or Bcl-xL[37]. In the present study, we found that nitrofen did not affect the expression of Bcl-2, Bax, and Bak (Figure 4A). However, the expression of Bcl-xL within nitrofen-treated cells was downregulated (Figure 4A), which was predicted to favor Bak and Bax homodimerization and apoptosis induction. Our findings also demonstrate a decline in the mitochondrial membrane potential after exposure to nitrofen (Figure 4B), indicating leakage of proteins from mitochondrial intermembrane space. AIF is a flavoprotein with activities of both oxidoreductase and DNA-binding domains, but no intrinsic DNase activity[38]. In the mitochondria, AIF is involved in cellular respiration[37] and is essential for cell survival[39]. Emerging evidence suggests that translocation of mitochondrial AIF into the nucleus is a hallmark of caspase-independent apoptosis[40]. When translocated to the nucleus, AIF binds the DNase Endo G, resulting in DNA fragmentation and cell death[41]. In this report, we demonstrate the nuclear translocation of AIF after treatment with nitrofen (Figure 4C), which provides obvious support for the contention that nitrofen is able to induce mitochondria-mediated caspase-independent apoptosis in A549 cells.
MAPK are phosphorylated in response to a series of extracellular stimuli. There are at least 3 subfamilies of the MAPK superfamily: ERK, JNK, and p38-MAPK[42], which have been implicated in both apoptosis and survival signaling. ERK are activated and play a critical role in transmitting signals initiated by growth factors, such as epidermal growth factor and platelet-derived growth factor[43]. JNK and p38-MAPK are potently activated by various forms of inflammatory signals or stress[44]. Previous studies demonstrated that the activation of MAPK was involved in ceramide-activated apoptotic signaling upstream of the mitochondria[45]. The p38-MAPK and ERK1/2 inhibitors attenuated the mitochondrial release of AIF[45]. In this study, we found that nitrofen administration resulted in the activation of p38-MAPK, but not of ERK1/2 or JNK (Figure 5A). This finding was similar to a recent report that found that nitrofen induced p38-MAPK activity in P19 teratocarcinoma cells, which was associated with reactive oxygen species[32]. Moreover, pharmacological inhibition of p38-MAPK in A549 cells was shown to abolish the nitrofen-induced downregula-tion of Bcl-xL, nuclear translocation of AIF, and apoptosis (Figure 5B,C), demonstrating that the activation of p38-MAPK is involved in nitrofen-induced apoptosis in A549 cells. However, at present, the involvement of other signaling cascades can not be ruled out, which warrants our further study.
In summary, in the present study we demonstrated that nitrofen treatment would decrease the proliferation of cultured A549 pneumocytes accompanied with the downregula-tion of PCNA. The inhibition of cell proliferation would relate to mitochondria-mediated caspase-independent apoptosis involving the activation of the p38-MAPK signaling pathway. These findings lay the groundwork for further investigation into the mechanisms of nitrofen-mediated pulmonary hypoplasia and for the characterization of the pathways involved in nitrofen-mediated apoptosis in type II pneumocytes.
Acknowledgements
We are grateful to Dr Dechun LI and Mr John LANGER (Johns Hopkins University School of Medicine, Baltimore, MD, USA) for their help in the preparation of this manuscript.
References
- Hurt SS, Smith JM, Hayes AW. Nitrofen: a review and perspective. Toxicology 1983;29:1-37.
- Manson JM. Mechanism of nitrofen teratogenesis. Environ Health Perspect 1986;70:137-47.
- Islam S, Narra V, Cote GM, Manganaro TF, Donahoe PK, Schnitzer JJ. Prenatal vitamin E treatment improves lung growth in fetal rats with congenital diaphragmatic hernia. J Pediatr Surg 1999;34:172-6.
- Alfonso LF, Vilanova J, Aldazabal P, Lopez de Torre B, Tovar JA. Lung growth and maturation in the rat model of experimentally induced congenital diaphragmatic hernia. Eur J Pediatr Surg 1993;3:6-11.
- Suen HC, Catlin EA, Ryan DP, Wain JC, Donahoe PK. Biochemical immaturity of lungs in congenital diaphragmatic hernia. J Pediatr Surg 1993;28:471-7.
- Alfanso LF, Arnaiz A, Alvarez FJ, Qi B, Diez-Pardo JA, Vallis-i-Soler A, et al. Lung hypoplasia and surfactant system immaturity induced in the fetal rat by prenatal exposure to nitrofen. Biol Neonate 1996;69:94-100.
- Coleman C, Zhao J, Gupta M, Buckley S, Tefft JD, Wuenschell CW, et al. Inhibition of vascular and epithelial differentiation in murine nitrofen-induced diaphragmatic hernia. Am J Physiol Lung Cell Mol Physiol 1998;274:L636-46.
- Cilley RE, Zgleszewski SE, Krummel TM, Chinoy MR. Nitrofen dose-dependent gestational day-specific murine lung hypoplasia and left-sided diaphragmatic hernia. Am J Physiol Lung Cell Mol Physiol 1997;272:L362-71.
- Chen MH, MacGowan A, Ward S, Bavik C, Greer JJ. The activation of the retinoic acid response element is inhibited in an animal model of congenital diaphragmatic hernia. Biol Neonate 2003;83:157-61.
- Mason RJ. Biology of alveolar type II cells. Respirology 2006;11:S12-5.
- Li J, Hu T, Liu W, Xiang B, Jiang X. Effect of epidermal growth factor on pulmonary hypoplasia in experimental diaphragmatic hernia. J Pediatr Surg 2004;39:37-42.
- Gonzalez-Reyes S, Martinez L, Tovar JA. Vitamin C rescues in part the effects of nitrofen on cultured human pneumocytes. Pediatr Surg Int 2004;20:258-62.
- Nardone LL, Andrews SB. Cell line A549 as a model of the type II pneumocyte. Phospholipid biosynthesis from native and organometallic precursors. Biochim Biophys Acta 1979;573:276-95.
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983;65:55-63.
- Sheffield LG. Effect of sialoadenectomy on the ability of mouse serum to induce deoxyribonucleic acid synthesis in mammary epithelial cells: possible role of epidermal growth factor. J Dairy Sci 1990;73:2087-92.
- Lebrin F, Goumans MJ, Jonker L, Carvalho RL, Valdimarsdottir G, Thorikay M, et al. Endoglin promotes endothelial cell proliferation and TGF-beta/ALK1 signal transduction. EMBO J 2004;23:4018-8.
- Overbeeke R, Steffens-Nakken H, Vermes I, Reutelingsperger C, Haanen C. Early features of apoptosis detected by four different flow cytometry assays. Apoptosis 1998;3:115-21.
- Leite M, Quinta-Costa M, Leite PS, Guimaraes JE. Critical evaluation of techniques to detect and measure cell death — study in a model of UV radiation of the leukaemic cell line HL60. Anal Cell Pathol 1999;19:139-51.
- Pietra G, Mortarini R, Parmiani G, Anichini A. Phases of apopto-sis of melanoma cells, but not of normal melanocytes, differently affect maturation of myeloid dendritic cells. Cancer Res 2001;61:8218-26.
- Cossarizza A, Baccarani-Contri M, Kalashnikova G, Franceschi C. A new method for the cytofluorimetric analysis of mitochondrial membrane potential using the J-aggregate forming lipophilic cation 5,5',6,6'-tetrachloro-1,1',3,3'-tetraethylbenzimi-dazolcarbocyanine iodide (JC-1). Biochem Biophys Res Commun 1993;197:40-5.
- Shrivastava A, Tiwari M, Sinha RA, Kumar A, Balapure AK, Bajpai VK, . Molecular iodine induces caspase-independent apoptosis in human breast carcinoma cells involving the mitochondria-mediated pathway. J Biol Chem 2006; 281: 19 762–71.
- Cass DL, Quinn TM, Yang EY, Liechty KW, Crombleholme TM, Flake AW, et al. Increased cell proliferation and decreased apoptosis characterize congenital cystic adenomatoid malformation of the lung. J Pediatr Surg 1998;33:1043-7.
- Casasco A, Giordano M, Danova M, Casasco M, Icaro Cornaglia A, Calligaro A. PC10 monoclonal antibody to proliferating cell nuclear antigen as probe for cycling cell detection in developing tissues. A combined immunocytochemical and flow cytometric study. Histochemistry 1993;99:191-9.
- Alles AJ, Losty PD, Donahoe PK, Manganaro TF, Schnitzer JJ. Embryonic cell death patterns associated with nitrofen-induced congenital diaphragmatic hernia. J Pediatr Surg 1995;30:353-60.
- Shao RG, Cao CX, Pommier Y. Activation of PKCalpha downstream from caspases during apoptosis induced by 7-hydro-xystaurosporine or the topoisomerase inhibitors, camptothecin and etoposide, in human myeloid leukemia HL60 cells. J Biol Chem 1997; 272: 31 321–5.
- Otera H, Ohsakaya S, Nagaura Z, Ishihara N, Mihara K. Export of mitochondrial AIF in response to proapoptotic stimuli depends on processing at the intermembrane space. EMBO J 2005;24:1375-86.
- Sarkar D, Su ZZ, Lebedeva IV, Sauane M, Gopalkrishnan RV, Valerie K, . mda-7 (IL-24) mediates selective apoptosis in human melanoma cells by inducing the coordinated overexpres-sion of the GADD family of genes by means of p38 MAPK. Proc Natl Acad Sci USA 2002; 99: 10 054–9.
- Porras A, Zuluaga S, Black E, Valladares A, Alvarez AM, Ambrosino C, et al. p38α mitogen-activated protein kinase sensitizes cells to apoptosis induced by different stimuli. Mol Biol Cell 2004;15:922-33.
- Maga G, Hubscher U. Proliferating cell nuclear antigen (PCNA): a dancer with many partners. J Cell Sci 2003;116:3051-60.
- Keijzer R, Liu J, Deimling J, Tibboel D, Post M. Dual-hit hypothesis explains pulmonary hypoplasia in the nitrofen model of congenital diaphragmatic hernia. Am J Pathol 2000;156:1299-306.
- Carvalho H, Evelson P, Sigaud S, Gonzalez-Flecha B. Mitogen-activated protein kinases modulate H(2)O(2)-induced apoptosis in primary rat alveolar epithelial cells. J Cell Biochem 2004;92:502-13.
- Kling DE, Aidlen JT, Fisher JC, Kinane TB, Donahoe PK, Schnitzer JJ. Nitrofen induces a redox-dependent apoptosis associated with increased p38 activity in P19 teratocarcinoma cells. Toxicol 2005;19:1-10.
- Bahi N, Zhang J, Llovera M, Ballester M, Comella JX, Sanchis D. Switch from caspase-dependent to caspase-independent death during heart development: essential role of endonuclease G in ischemia-induced DNA processing of differentiated cardio-myocytes. J Biol Chem 2006; 281: 22 943–52.
- McBurney MW, Jones-Villeneuve EM, Edwards MK, Anderson PJ. Control of muscle and neuronal differentiation in a cultured embryonal carcinoma cell line. Nature 1982;299:165-7.
- Ravagnan L, Roumier T, Kroemer G. Mitochondria, the killer organelles and their weapons. J Cell Physiol 2002;192:131-7.
- Griffiths GJ, Dubrez L, Morgan CP, Jones NA, Whitehouse J, Corfe BM, et al. Cell damage-induced conformational changes of the pro-apoptotic protein bak in vivo precede the onset of apoptosis. J Cell Biol 1999;144:903-14.
- Yin XM, Oltvai ZN, Korsmeyer SJ. BH1 and BH2 domains of Bcl-2 are required for inhibition of apoptosis and heterodimeriza-tion with Bax. Nature 1994;369:321-3.
- Daugas E, Nochy D, Ravagnan L, Loeffler M, Susin SA, Zamzami N, et al. Apoptosis-inducing factor (AIF): a ubiquitous mitochondrial oxidoreductase involved in apoptosis. FEBS Lett 2000;476:118-23.
- Klein JA, Longo-Guess CM, Rossmann MP, Seburn KL, Hurd RE, Frankel WN, et al. The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature 2002;419:367-74.
- Cande C, Cohen I, Daugas E, Ravagnan L, Larochette N, Zamzami N, et al. Apoptosis-inducing factor (AIF): a novel caspase-independent death effector released from mitochondria. Biochimie 2000;84:215-22.
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999;397:441-6.
- Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene 2004;23:2838-49.
- Seger R, Krebs EG. The MAPK signaling cascade. FASEB J 1995;9:726-35.
- Irigoyen JP, Besser D, Nagamine Y. Cytoskeleton reorganization induces the urokinase-type plasminogen activator gene via the Ras/extracellular signal-regulated kinase (ERK) signaling pathway. J Biol Chem 1997;272:1904-9.
- Stoica BA, Movsesyan VA, Knoblach SM, Faden AI. Ceramide induces neuronal apoptosis through mitogen-activated protein kinases and causes release of multiple mitochondrial proteins. Mol Cell Neurosci 2005;29:355-71.