
Time-dependence of cardiomyocyte differentiation disturbed by peroxisome proliferator-activated receptor α inhibitor GW6471 in murine embryonic stem cells in vitro1
Introduction
Peroxisome proliferator-activated receptor α (PPARα) is a member of the nuclear receptor superfamily of transcription factors and binds cognate response elements as an obligate heterodimer with the retinoid X receptor. PPARα plays a key role in the transcriptional regulation of genes encoding mitochondrial fatty acid β-oxidation (FAO) enzymes during cardiac development and in response to physiological and pathophysiological stimuli, including fasting, cardiac hypertrophy, and cellular hypoxia[1-4]. FAO enzymes, including medium chain acyl co-enzyme A, dehydrogenase, and muscle carnitine palmitoyltransferase I (M-CPT I or CPT Iβ)[5–7], are the principal sources of energy production in adult mammalian cardiomyocytes. PPARα is rich in tissues that have high energy demand, such as heart, skeletal muscle, brown fat, kidney, liver, and brain[8], demonstrating a close association between PPARα and energy turnover.
However, little is known about how physiological or pathophysiological signals are transduced for the modulation of the transcription of PPARα in cardiomyocytes. The p38 mitogen-activated protein kinase (MAPK) signaling pathway could be activated by cellular stressors in the heart, including ischemia, hypoxia, and hypertrophic growth stimuli[9]. As mentioned earlier, the expression of PPARα is modulated to adapt to different demands in these processes. Therefore, we hypothesize that p38 MAPK may act as the upstream event that regulates the transcription of PPARα.
PPARα, together with PPARδ and PPARγ, form a subgroup within the nuclear receptor superfamily[8,10]. PPAR isoforms have been demonstrated to be involved in the differentiation of several cell types, including nerve cells, adipose cells, and some tumor cell types[11–13]. Prominent increased mRNA of PPARα has been observed in the heart during embryonic development starting on d 7 in vivo[14,15]. This prominent expression abates before birth, suggesting that a period of PPARα exposure may be critical to the normal development of the heart.
It is increasingly clear that the p38 MAPK pathway plays an important role in a large number of cellular processes, such as cell growth, cell differentiation, cell cycle arrest, and apoptosis[9,16–20]. In mice, p38 MAPK activity was recently demonstrated to be required for the development of the 8–16 cell stage embryos[17]. In the mouse embryonic carcinoma cell model, the activation of p38 MAPK has been shown to be necessary for cardiac differentiation[16,21].
Although multiple roles for PPARα have been proposed, little is known of the significance of PPARα in early cardiac development, especially during the differentiation of cardiomyocytes. Using embryonic stem (ES) cells as a model system faithfully recapitulates cardiomyocyte differentiation[22–24]. The present study was designated to determine the possible function of PPARα in early cardiomyocyte differentiation of ES cells in vitro. In addition, the p38 MAPK pathway was also detected to evaluate the possible pathway regulating the transcription of PPARα.
Materials and methods
Cell culture and differentiation The permanent ES cell line D3 (American Type Culture Collection, CRL-1934, Manassas, VA, USA) was cultivated in undifferentiated states on primary cultures of mouse embryonic fibroblasts in Dulbecco’s modified Eagle’s minimal essential medium (DMEM, Gibco BRL, Life Technologies, Germany), supplemented with 10% fetal calf serum (FCS, Gibco, Germany), 1×10-4 mol/L beta-mercaptoethanol (Sigma, St Louis, MO, USA), non-essential amino acids (NEAA, Hyclone, Logan, UT, USA, stock solution dilution 1:100), and 1×106 units/L recombinant mouse leukemia inhibitory factor (Chemicon, Temecula, California, USA). For the differentiation of ES cells, embryoid bodies (EB) were generated using the hanging drop method[25,26]. On d 0, 30 microlitres of drops containing approximately 600 ES cells were placed on the lids of Petri dishes filled with D-Hanks’ solution, and cultivated in hanging drops for 3 d followed by another 2 d in the Petri dishes. On d 5, the EB were plated separately onto gelatin-coated 24-well culture plates in differentiation medium that consisted of DMEM, 20% FCS, 1 µmol/L mercaptoethanol, and 1% NEAA. GW6471 and SB203580 were added, respectively, at the time points indicated in the text.
Reagents SB203580 was purchased from Biomol (Plymouth Meeting, PA, USA). GW6471 was obtained from Sigma-Aldrich (USA). Each inhibitor was dissolved in DMSO at 1000×immediately prior to use. Unused inhibitor was aliquoted into Eppendorf tubes and stored at -20 °C.
RT-PCR The total RNA was isolated from the ES cells and EB using Trizol reagent (Gibco BRL, Germany) in accordance with the manufacturer’s instructions. To synthesize first strand cDNA, 1 µg total RNA was incubated with 0.5 µg of oligo (dT) 6 primer (Sangon, Shanghai, China) and 5 µL deionized water at 65 °C for 15 min. Reverse transcription reactions of 20 µL were performed with 200 units of M-MuLV reverse transcriptase (Gibco BRL, Germany), 4 µL of 5×reaction buffer, and 1 mmol/L deoxynucleoside triphosphate (dNTP) mixture for 1 h at 42 °C. Polymerase chain reactions of 50 µL contained 1 µL of the RT reaction product, 5 µL of 10×PCR buffer, 25 units Taq polymerase (Sangon, China), 1 µL of 10 mmol/L dNTP mixture, and 30 pmol of each primer.
Primers, annealing temperature, product size, and the number of PCR cycles are depicted in Table 1. Products were analyzed in 1.5% agarose, ethidium bromide staining gels. β-actin was used as an internal standard. No PCR products were found without RT reaction and in water controls. RNA from 3 independent experiments were analyzed.

Full table
Western blot analysis The cells were washed with phosphate-buffered saline PBS, collected in radioimmuno-precipitation assay RIPA buffer (containing 0.2% Triton X-100, 5 mmol/L EDTA, 1 mmol/L phenylmethanesulfonyl fluoride PMSF, 0.01 g/L leupeptin, and 0.01 g/L aprotinin) and lysed for 30 min on ice. Aliquots were assayed for protein concentration using the Bio-Rad protein assay kit (Hercules, CA, USA); equal amounts of proteins were loaded per well on a 12% SDS-PAGE. Subsequently, the proteins were transferred onto 0.45 µm pore size nitrocellulose membranes and blocked with 5% dry milk in PBS (pH 7.4, with 0.1% Tween 20) at room temperature. The blots were probed with either rabbit polyclonal anti-PPARα, mouse monoclonal anti-troponin-T, goat polyclonal anti-actin (dilution 1:500, Santa Cruz Biotechnology, Santa Cruz, CA, USA), mouse monoclonal anti-α-actinin (dilution 1:500, Sigma-Aldrich, USA), rabbit polyclonal anti-phospho-p38 MAPK or rabbit polyclonal anti-p38 MAPK (dilution 1:1000, Cell Signaling Technology, USA) antibodies overnight at 4 °C, followed by washing 3 times with PBS-Tween (0.1% Tween 20) at room temperature and challenged with horseradish peroxidase HRP-conjugated goat anti-rabbit, rabbit anti-goat, or mouse anti-mouse antibodies (dilution 1:4000, Affinity Bioreagents, Golden, CO, USA), respectively. The proteins were visualized autoradiographi-cally with an enhanced chemiluminescent substrate (Pierce, Rockford, IL, USA) and scanned using a bio-imaging analyzer (Bio-Rad, USA).
Immunofluorescence analysis Differentiated EB that had been grown on coverslips were fixed for 20 min in methanol at -20 °C, followed by permeabilization in 0.1% Tween 20 in PBS. After washing in PBS three times, the EB on the coverslips were transferred to PBS containing 10% goat serum for 30 min at room temperature. The EB were then placed into blocking buffer containing mouse monoclonal anti-α-actinin antibodies (dilution 1:100) and incubated overnight at 4 °C, followed by incubation in blocking buffer containing fluorescein isothiocyanate (FITC)-labeled mouse anti-mouse antibody (1:500, Sigma-Aldrich, USA). For double staining, rabbit polyclonal anti-PPARα and mouse monoclonal anti-troponin-T were added together with the last dilution of 1:100, followed by incubation in blocking buffer containing FITC-labeled mouse anti-mouse (1:250, Sigma-Aldrich, USA) and Texa red-labeled goat anti-rabbit (1:250, Santa Cruz Biotechnology, USA) antibodies. For the morphometric analysis for the expression of sarcomeric proteins, all aspects of cell processing, immunostaining, and imaging were rigorously standardized. Digital images were obtained from randomly selected fields using 10×objective lens and analyzed by the image analysis software of the confocal setup (Leica TCS SP2, Bensheim, Germany). For fluorescence excitation, the 488 nm band of the argon ion laser of the confocal setup was used. Emission was recorded using a longpass LP505-nm filter set (Leica, Germany). Data are expressed as mean±SD calculated as a percentage of the cells in the controls. At least 5 different fields were measured for each dish.
Statistical analysis Student’s t-test and ANOVA were used to determine the statistical significance of differences between the values for the various experimental and control groups. P<0.05 was considered statistically significant.
Results
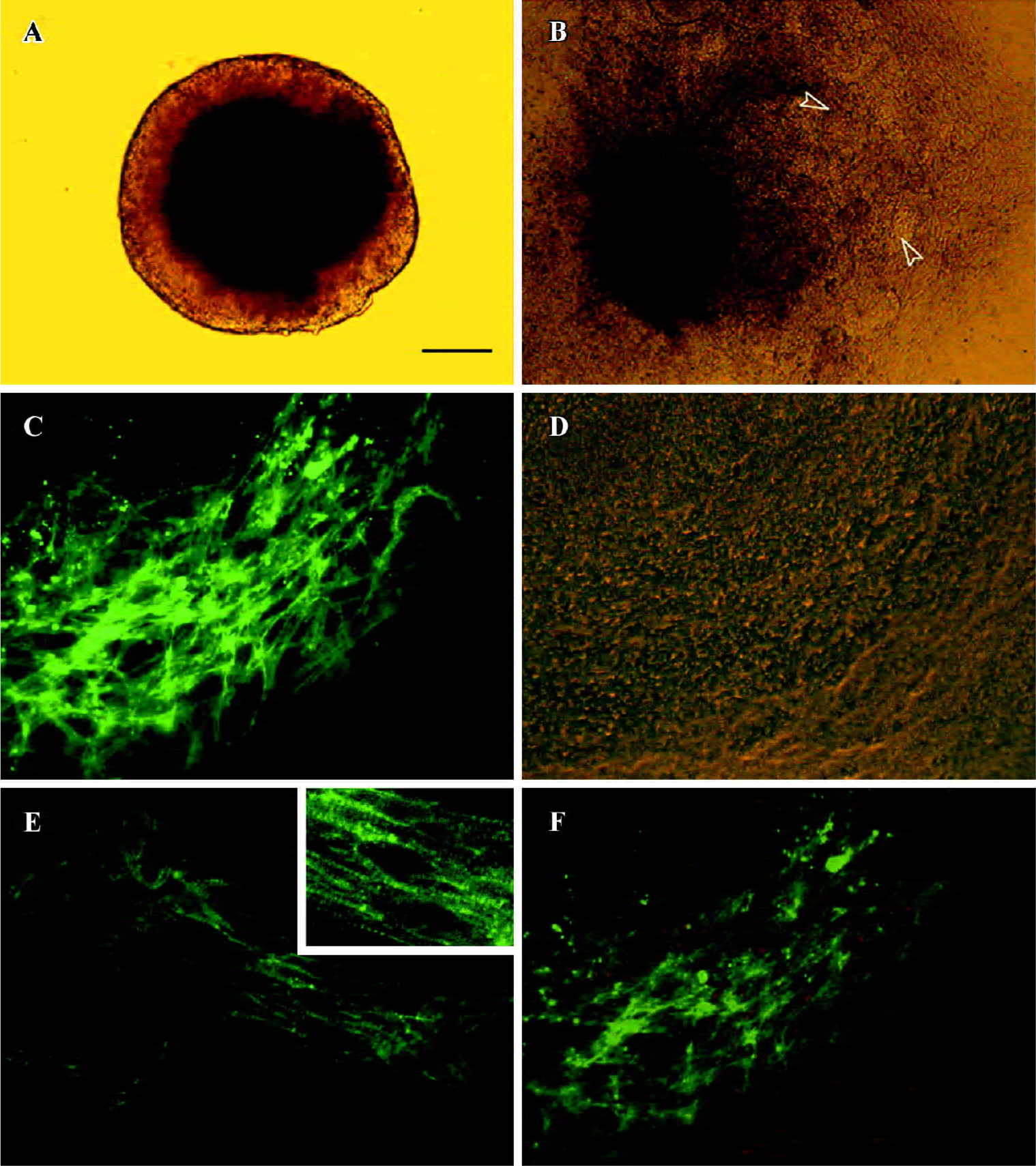
In vitro cardiomyocyte differentiation of ES cells The study protocol of cardiac differentiation of murine ES cells in vitro are shown in Figure 1, indicating the time points for RT-PCR, Western blotting, and immunofluorescence. The attached culture was established by plating a single, d 5 EB (Figure 2A) onto a 24-well plate and allowing continued cellular proliferation and differentiation. Within this multicellular arrangement in EB outgrowth, cardiomyocytes appeared as spontaneously contracting, round cell clusters. Each EB contained 1 or more beating areas (Figure 2B, 5 d after plating). An increase in size, strength of contraction, and beat frequency was observed during further differentiation. The synchronously contracting cardiomyocytes were positive for the anti-troponin-T antibody (Figure 2C). At higher magnification, cross striations were demonstrated with α-actinin immunolabeling (Figure 2E).
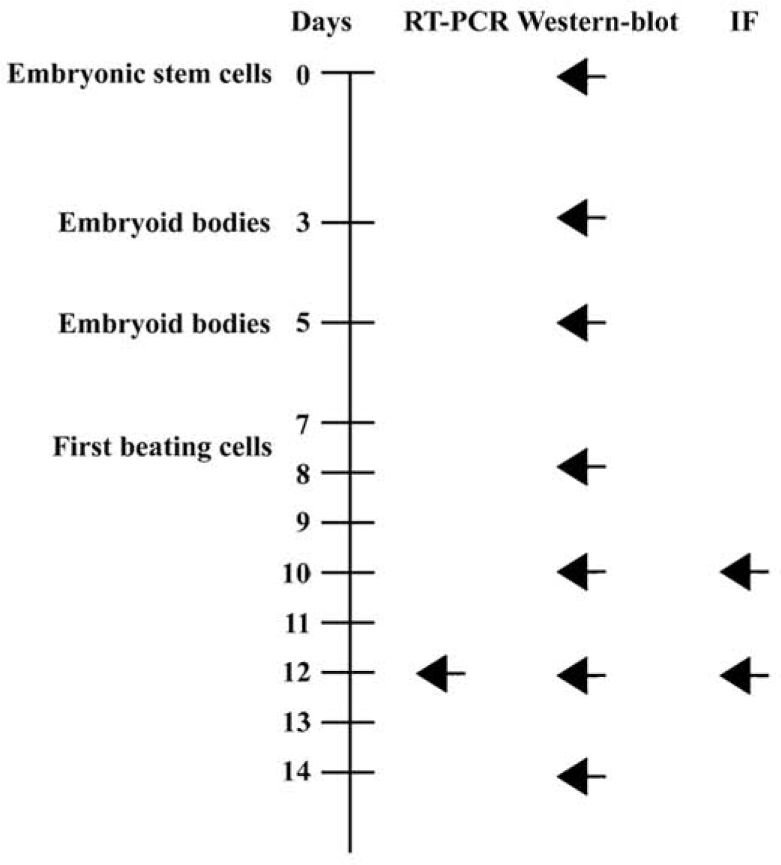
PPARα increased during cardiomyocyte differentiation Differentiation of ES cells into cardiomyocytes first required their forming into EB by growing them in hanging drop cultures. After 3 d in hanging drops, the EB were placed into suspension for 2 d, after which they were plated into individual wells of a 24-well plate. Beating commenced in distinct areas within EB on average 2 d later (culture d 7), the area of contracting cells expanded multifocally thereafter and reached a plateau around d 12. Therefore, these 2 time points were chosen for future analysis. As determined by Western blotting, the expression of PPARα was at low levels in early differentiation, but increased obviously in parallel with the appearance of contracting cardiomyocytes (Figure 3A).
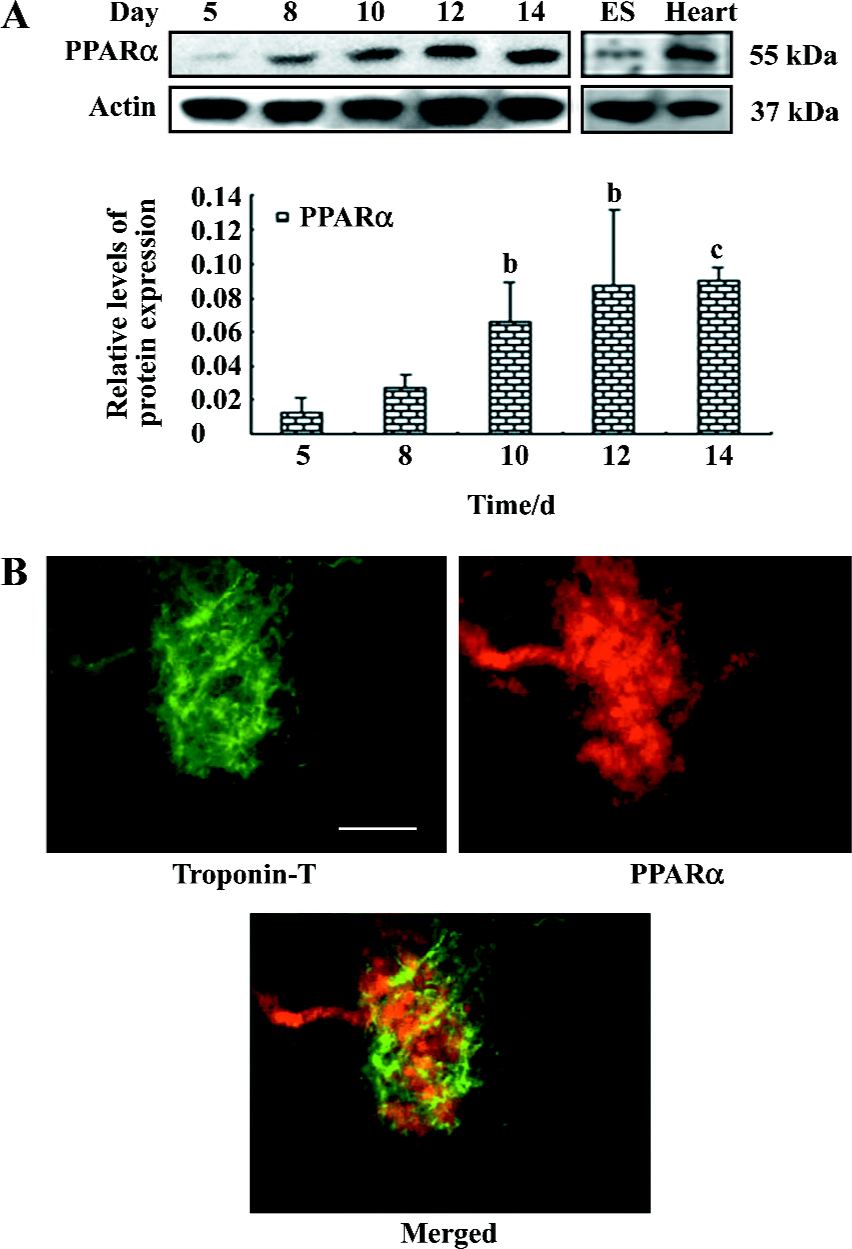
To confirm the result of the protein expression found by Western blotting, cultures were fixed at d 10 and doubly stained with antibodies against PPARα and cardiac troponin-T. In each case, we selected fields which were full of cells so that a comparison could be made between the level of staining in the cells positive for the marker and those that were negative. There was a strong correlation between PPARα and troponin-T during differentiation; a high level of PPARα was observed in cardiomyocytes positive for troponin-T (Figure 3B). Negative areas were also positive for PPARα, but the staining was at considerably low levels.
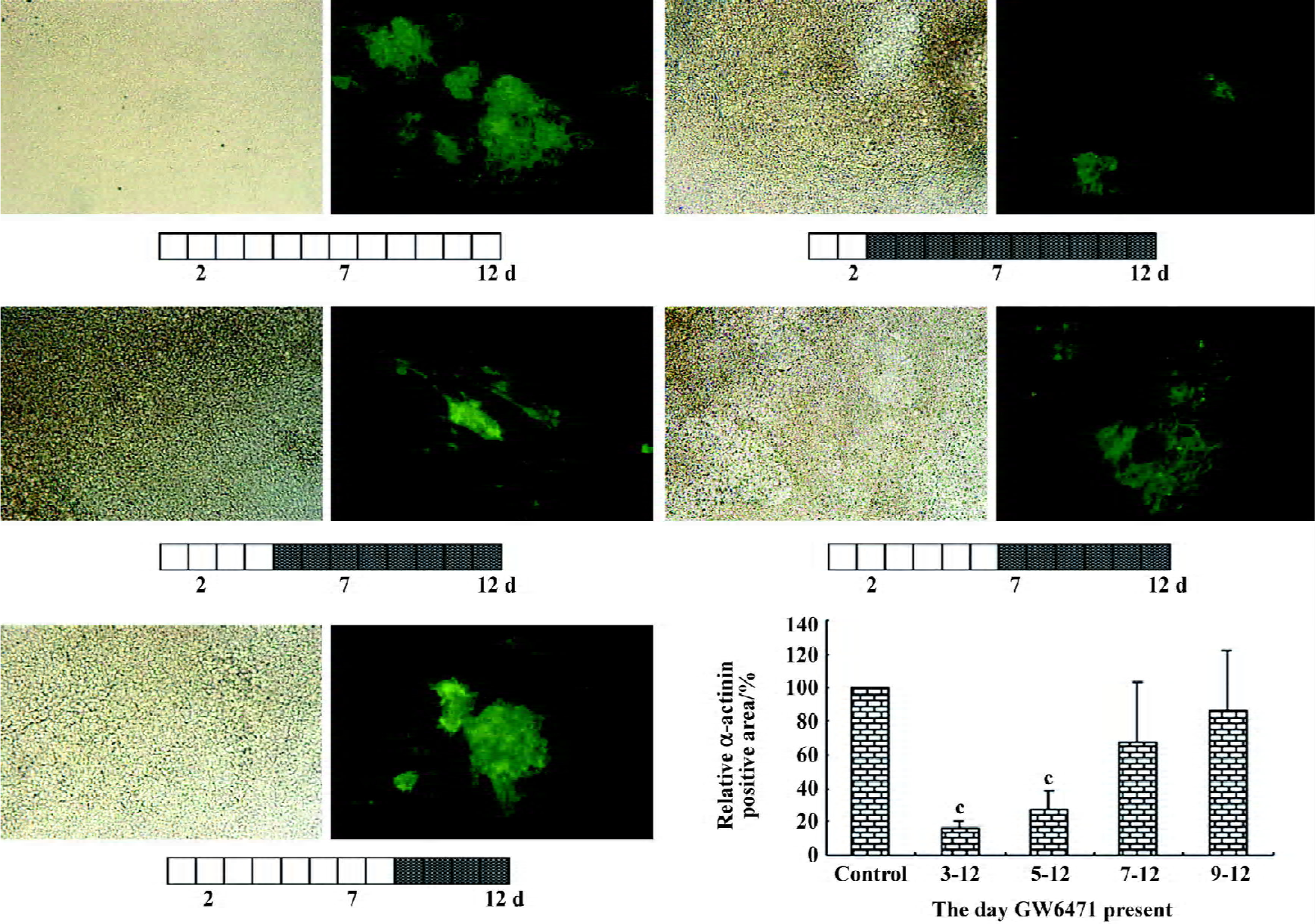
Specific inhibition of PPARα prevented cardiomyocyte differentiation of ES cells in a time-dependent manner GW6471, the specific inhibitor of PPARα[27], was used in our experiment to address whether these changes in the PPARα expression was a causative or a simple effect resulting from beating area formation. To closely define the exact time point during the course of the developmental program at which PPARα activity was essential, GW6471 was applied at different time courses of differentiation. The data showed that only the application of GW6471 before d 9 could efficiently prevent EB from cardiomyocyte differentiation, indicated by the reduced formation of the beating area (Figure 4) and expression of α-actinin and troponin-T (Figure 5B). When GW6471 was applied after d 9, cardiomyocyte differentiation and formation was slightly affected.
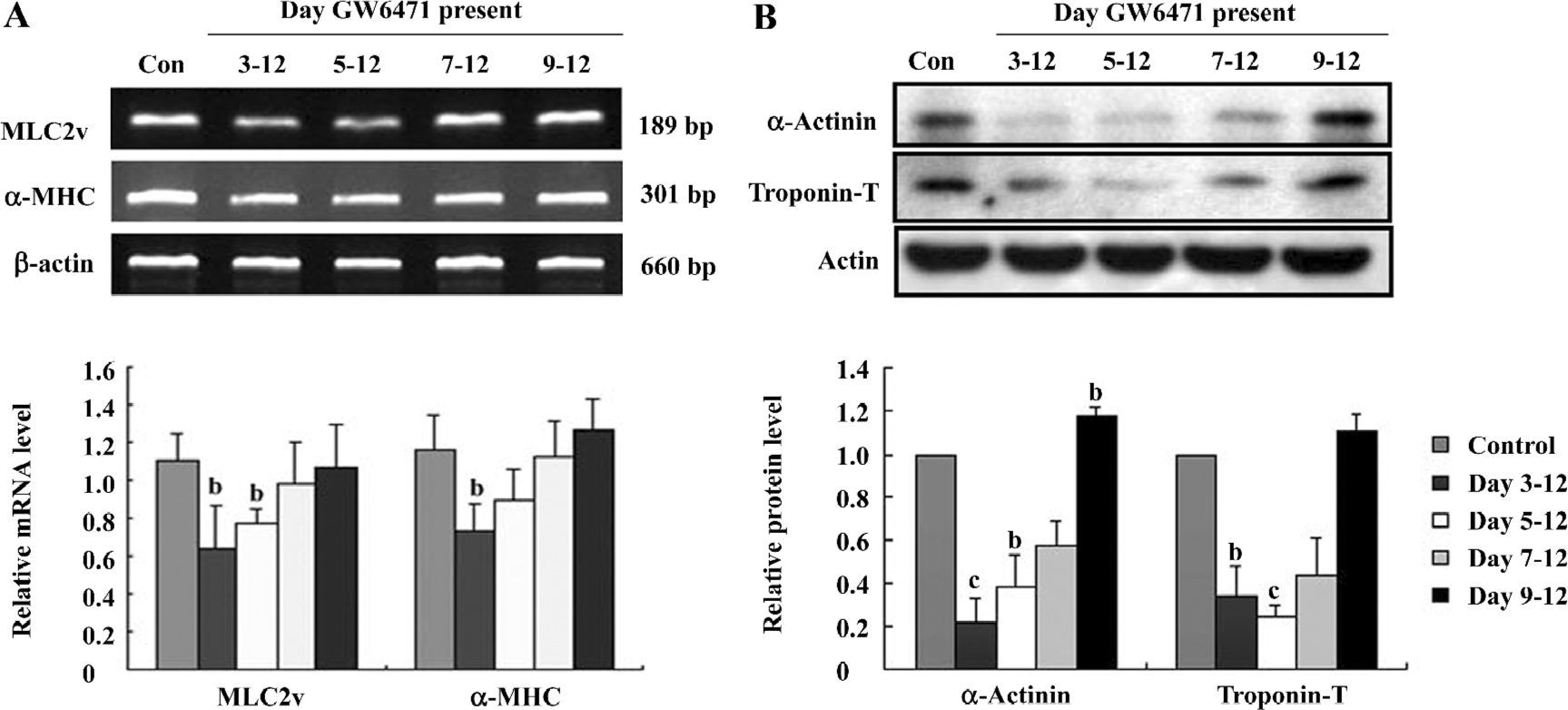
The expressions of α-MHC and MLC2v were analyzed by semiquantitative RT-PCR as they were specific transcription factors in cardiomyocyte differentiation. As a result, GW6471 tended to reduce the concentration of α-MHC and MLC2v compared with the appropriate controls (Figure 5A).
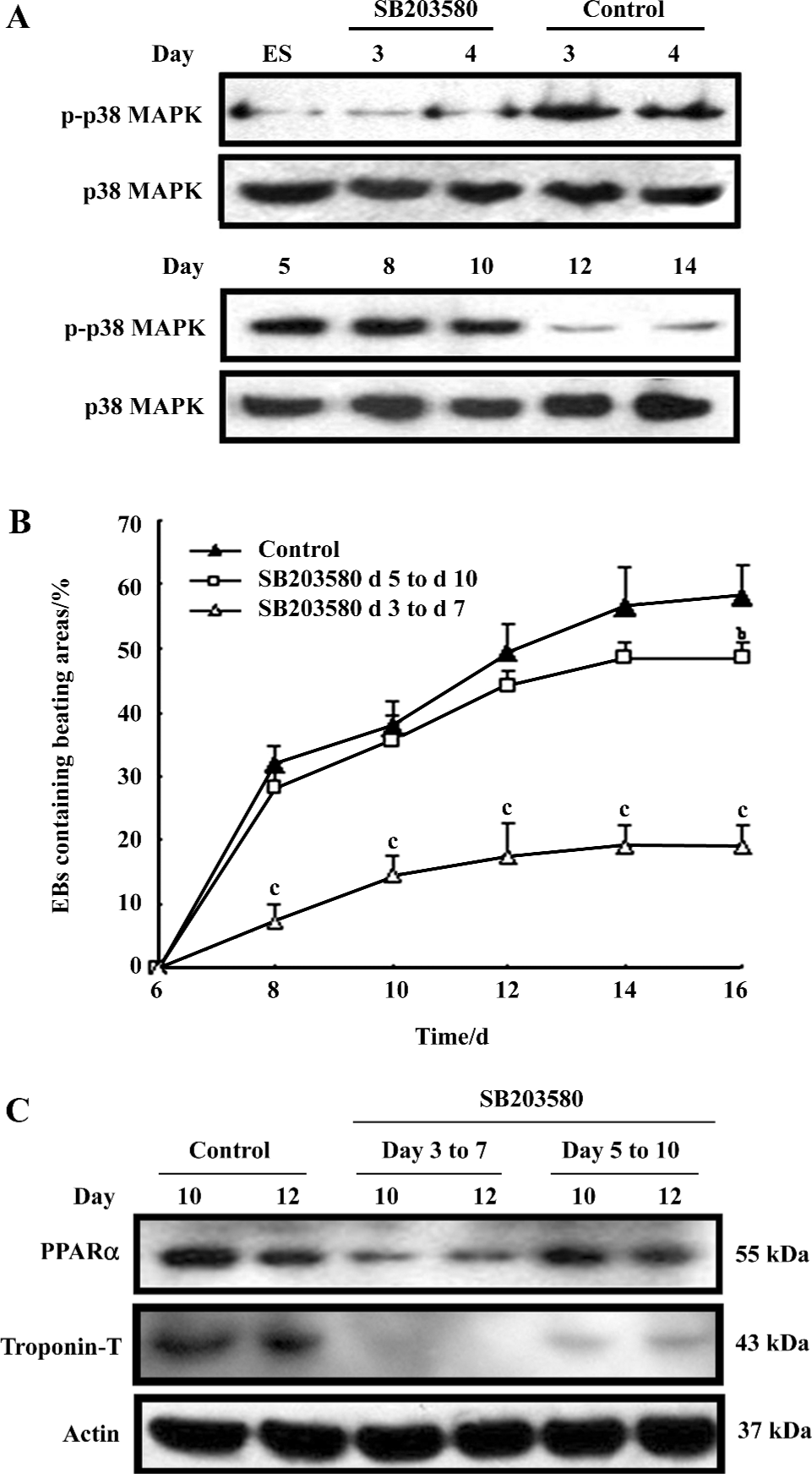
Inhibition of p38 MAPK prevented cardiomyocyte differentiation and reduced the expression of PPARα To explore the function of p38 MAPK in cardiomyocyte differentiation and the expression of PPARα, SB203580, the specific inhibitor of p38 MAPK, was employed in our experiment. P-p38 MAPK activity was maintained at a high level from d 3 and was followed by a decrease on d 10 (Figure 6A). The specific inhibition of p38 MAPK from d 3 to d 7 greatly reduced the formation of beating cardiomyocytes in the EB (Figure 6B) which was well matched with expression of PPARα (Figure 6C). While treated with SB203580 from d 5, cardiac differentiation and PPARα expression were just slightly affected. The expression of troponin-T was analyzed to confirm the inhibition of differentiation which was completely absent from the undifferentiated cells and the cells treated with SB203580 from d 3 to d 7. These results implied that the upregulation of PPARα was restrained to cardiac differentiation of ES cells in vitro, and p38 MAPK might be the signal pathway regulating the expression of PPARα.
Discussion
In this investigation, we gain insight into the expression and possible function of PPARα during the cardiomyocyte differentiation of ES cells in vitro. PPARα was observed to increase immediately after beating area formation. Interestingly, this phenomenon was recently replicated as the mRNA levels of PPARα increased almost 10-fold during myogenesis[28].
Cardiomyocyte differentiation can be divided into two processes: cardiogenesis and cardiac myofibrillogenesis. At d 3, cardiac transcription factors (ie GATA4, NKX2.5, MEF2C) are not expressed yet, while on d 5, cardiac transcription factors start to be fully expressed, but those encoding sarcomeric proteins are not. Since d 5 is a critical window at which the cardiac differentiation program becomes fully activated, but no cardiac cells identifiable by organized cardiac sarcomeric proteins (ie α-MHC, MLC2V) are yet present[29], PPARα appeared to mainly play a role in myofibrillogenesis. Our data showed that the inhibition of PPARα before d 9 significantly disrupted differentiation and reduced mRNA levels of α-MHC and MLC2v.
Mitochondrial number and functional capacity are dynamically regulated in accordance with cardiac energy demands during developmental stages and in response to diverse physiological conditions[30]. Since the cardiomyocyte differentiation of ES cells in vitro faithfully replicates the process in vivo, and these cardiomyocytes display properties similar to those observed in those in vivo or in primary cultures, there must be an increase in mitochondrial number with the emergence of beating clusters. In addition to the activation of suites of genes encoding contractile proteins, cardiac differentiation is accompanied by an organization of metabolism. Therefore, the most obvious explanation for our findings was that the inhibition of PPARα impaired mitochondria ATP production or function of mitochondria, thus hampering normal development. That blockade of mitochondrial activity by the inhibition of mitochondrial protein synthesis, uncoupling of the inner membrane potential from ATP synthesis, and the inhibition of mitochondrial ATP production inhibits differentiation of skeletal muscle cells from myoblast precursors[31]. The other explanation for our result may be that PPARα was involved in the regulation of the nitric oxid (NO)/nitric oxid synthase (NOS) system and thus affected cardiac differentiation. The beneficial effects of the activation of PPARα in the improvement of cardiovascular function have long been reported and these beneficial effects in cardiovascular diseases have been suggested to involve the NO/NOS system[32,33]. Interestingly, NO signaling plays an important role in cardiac differentiation and NO treatment can accelerate mouse ES cells to differentiate into cardiomyocyte by both inducing a switch toward a cardiac phenotype and inducing apoptosis in cells not committed to cardiac differentiation[34].
p38 MAPK was involved in cardiomyocyte differentiation of mouse embryonic carcinoma cells. Recently, p38 MAPK was further demonstrated to commit ES cells to cardiomyogenesis[18]. Consistent with these previous studies, p-p38 MAPK was found to maintain at a high level from d 3 to d 10, which overlaps with the increase of PPARα. Furthermore, the inhibition of p38 MAPK from d 3 to d 7 significantly prevented cardiac differentiation and reduced the expression PPARα, which implied a relationship between PPARα and p38 MAPK.
A previous study revealed that ligand-activated PPARα would drive its own transcription[35], and this effect would be increased if PPARα was phosphorylated by p38 MAPK[36]. There would have been abundant natural ligands (eg fatty acids) for the activation of PPARα since EB were grown in the presence of 20% fetal bovine serum during differentiation. p38 MAPK-mediated phosphorylation activates PPARα in a ligand-influenced manner and results in enhanced functional cooperation with the transcriptional co-activator peroxisome proliferator-activated receptor γ coactivator (PGC)-1α. Although we have no detailed experimental data on the mechanism by which p38 MAPK mediates the expression of PPARα, two possibilities can be considered. First, p38 MAPK may enhance the binding between PPARα and PGC-1α and thus stimulates the transcription of PPARα. Second, the activation of p38 MAPK can improve the stability of PPARα during differentiation and thus contributes to the increase of PPARα. However, we could not exclude the possibility that other upstream proteins might be responsible for the mediation of PPARα during cardiac differentiation in vitro.
In conclusion, PPARα increased in cardiomyocyte differentiation in vitro, and p38 MAPK partly acted as the upstream event regulating the expression of PPARα. The inhibition of PPARα in the early course markedly prevented cardiac differentiation, as indicated by the reduced expression of cardiac sarcomeric proteins and specific genes.
References
- Depre C, Shipley GL, Chen W, Han Q, Doenst T, Moore ML, et al. Unloaded heart in vivo replicates fetal gene expression of cardiac hypertrophy. Nat Med 1998;4:1269-75.
- Leone TC, Weinheimer CJ, Kelly DP. A critical role for the peroxisome proliferator-activated receptor alpha (PPARalpha) in the cellular fasting response: the PPARalpha-null mouse as a model of fatty acid oxidation disorders. Proc Natl Acad Sci USA 1999;96:7473-8.
- Sack MN, Rader TA, Park S, Bastin J, McCune SA, Kelly DP. Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circulation 1996;94:2837-42.
- Sack MN, Disch DL, Rockman HA, Kelly DP. A role for Sp and nuclear receptor transcription factors in a cardiac hypertrophic growth program. Proc Natl Acad Sci USA 1997;94:6438-43.
- Brandt JM, Djouadi F, Kelly DP. Fatty acids activate transcription of the muscle carnitine palmitoyltransferase I gene in cardiac myocytes via the peroxisome proliferator-activated receptor alpha. J Biol Chem 1998;273:23786-92.
- Mascaro C, Acosta E, Ortiz JA, Marrero PF, Hegardt FG, Haro D, et al. Control of human muscle-type carnitine palmitoyltrans-ferase I gene transcription by peroxisome proliferator-activated receptor. J Biol Chem 1998;273:8560-3.
- Yu GS, Lu YC, Gulick T. Co-regulation of tissue-specific alternative human carnitine palmitoyltransferase Ibeta gene promoters by fatty acid enzyme substrate. J Biol Chem 1998;273:32901-9.
- Kliewer SA, Forman BM, Blumberg B, Ong ES, Borgmeyer U, Mangelsdorf DJ, et al. Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc Natl Acad Sci USA 1994;91:7355-9.
- Ono K, Han J. The p38 signal transduction pathway: activation and function. Cell Signal 2000;12:1-13.
- Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 1990;347:645-50.
- Rosen ED, Sarraf P, Troy AE, Bradwin G, Moore K, Milstone DS, et al. PPAR gamma is required for the differentiation of adipose tissue in vivo and in vitro. Mol Cell 1999;4:611-7.
- Mueller E, Sarraf P, Tontonoz P, Evans RM, Martin KJ, Zhang M, et al. Terminal differentiation of human breast cancer through PPAR gamma. Mol Cell 1998;1:465-70.
- Park KS, Lee RD, Kang SK, Han SY, Park KL, Yang KH, et al. Neuronal differentiation of embryonic midbrain cells by upregulation of peroxisome proliferator-activated receptor-gamma via the JNK-dependent pathway. Exp Cell Res 2004;297:424-33.
- Steinmetz M, Quentin T, Poppe A, Paul T, Jux C. Changes in expression levels of genes involved in fatty acid metabolism: upregulation of all three members of the PPAR family (alpha, gamma, delta) and the newly described adiponectin receptor 2, but not adiponectin receptor 1 during neonatal cardiac development of the rat. Basic Res Cardiol 2005;100:263-9.
- Braissant O, Wahli W. Differential expression of peroxisome proliferator-activated receptor-alpha, -beta, and -gamma during rat embryonic development. Endocrinology 1998;139:2748-54.
- Davidson SM, Morange M. Hsp25 and the p38 MAPK pathway are involved in differentiation of cardiomyocytes. Dev Biol 2000;218:146-60.
- Natale DR, Paliga AJ, Beier F, D’Souza SJ, Watson AJ. p38 MAPK signaling during murine preimplantation development. Dev Biol 2004;268:76-88.
- Aouadi M, Bost F, Caron L, Laurent K, Le Marchand Brustel Y, Binetruy B, et al. p38 mitogen-activated protein kinase activity commits embryonic stem cells to either neurogenesis or cardiomyogenesis. Stem Cells 2006;24:1399-406.
- Oskouian B, Sooriyakumaran P, Borowsky AD, Crans A, Dillard-Telm L, Tam YY, et al. Sphingosine-1-phosphate lyase potentiates apoptosis via p53- and p38-dependent pathways and is down-regulated in colon cancer. Proc Natl Acad Sci USA 2006;103:17384-9.
- Cai B, Chang SH, Becker EB, Bonni A, Xia Z. p38 MAP kinase mediates apoptosis through phosphorylation of BimEL at Ser-65. J Biol Chem 2006;281:25215-22.
- Eriksson M, Leppa S. Mitogen-activated protein kinases and activator protein 1 are required for proliferation and cardio-myocyte differentiation of P19 embryonal carcinoma cells. J Biol Chem 2002;277:15992-6001.
- Wobus AM, Wallukat G, Hescheler J. Pluripotent mouse embryonic stem cells are able to differentiate into cardiomyocytes expressing chronotropic responses to adrenergic and cholinergic agents and Ca2+ channel blockers. Differentiation 1991;48:173-82.
- Metzger JM, Lin WI, Samuelson LC. Vital staining of cardiac myocytes during embryonic stem cell cardiogenesis in vitro. Circ Res 1996;78:547-52.
- Hescheler J, Fleischmann BK, Lentini S, Maltsev VA, Rohwedel J, Wobus AM, et al. Embryonic stem cells: a model to study structural and functional properties in cardiomyogenesis. Cardiovasc Res 1997;36:149-62.
- Metzger JM, Lin WI, Johnston RA, Westfall MV, Samuelson LC. Myosin heavy chain expression in contracting myocytes isolated during embryonic stem cell cardiogenesis. Circ Res 1995;76:710-9.
- Scholz G, Pohl I, Genschow E, Klemm M, Spielmann H. Embryotoxicity screening using embryonic stem cells in vitro: correlation to in vivo teratogenicity. Cells Tissues Organs 1999;165:203-11.
- Xu HE, Stanley TB, Montana VG, Lambert MH, Shearer BG, Cobb JE, et al. Structural basis for antagonist-mediated recruitment of nuclear co-repressors by PPARalpha. Nature 2002;415:813-7.
- Kraft CS, LeMoine CM, Lyons CN, Michaud D, Mueller CR, Moyes CD, et al. Control of mitochondrial biogenesis during myogenesis. Am J Physiol Cell Physiol 2006;290:C1119-27.
- Wei H, Juhasz O, Li J, Tarasova YS, Boheler KR. Embryonic stem cells and cardiomyocyte differentiation: phenotypic and molecular analyses. J Cell Mol Med 2005;9:804-17.
- Attardi G, Schatz G. Biogenesis of mitochondria. Annu Rev Cell Biol 1988;4:289-333.
- Herzberg NH, Middelkoop E, Adorf M, Dekker HL, Van Galen MJ, Van den Berg M, et al. Mitochondria in cultured human muscle cells depleted of mitochondrial DNA. Eur J Cell Biol 1993;61:400-8.
- Frick MH, Elo O, Haapa K, Heinonen OP, Heinsalmi P, Helo P, et al. Helsinki Heart Study: primary-prevention trial with gemfibrozil in middle-aged men with dyslipidemia. Safety of treatment, changes in risk factors, and incidence of coronary heart disease. N Engl J Med 1987;317:1237-45.
- Rubins HB, Robins SJ, Collins D, Fye CL, Anderson JW, Elam MB, et al. Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial Study Group. N Engl J Med 1999;341:410-8.
- Kanno S, Kim PK, Sallam K, Lei J, Billiar TR, Shears LL II. Nitric oxide facilitates cardiomyogenesis in mouse embryonic stem cells. Proc Natl Acad Sci USA 2004;101:12277-81.
- Pineda Torra I, Jamshidi Y, Flavell DM, Fruchart JC, Staels B. Characterization of the human PPARalpha promoter: identification of a functional nuclear receptor response element. Mol Endocrinol 2002;16:1013-28.
- Barger PM, Browning AC, Garner AN, Kelly DP. p38 mitogen-activated protein kinase activates peroxisome proliferator-activated receptor alpha: a potential role in the cardiac metabolic stress response. J Biol Chem 2001;276:44495-501.