
A cell-based model of α-synucleinopathy for screening compounds with therapeutic potential of Parkinson’s disease1
Introduction
Parkinson’s disease (PD), characterized pathologically by the selective loss of dopaminergic neurons and the presence of Lewy bodies (LB) in the substantia nigra, is the second most common neurodegenerative disorder after Alzheimer’s disease, affecting people in mid and late life[1]. Mutations of the parkin and α-synuclein genes have been associated with autosomal recessive and dominant parkinsonism pedigrees, respectively[2]. α-Synuclein is a highly-conserved, 140 amino acid protein which is mainly expressed in neurons and concentrated in presynaptic nerve terminals[3,4]. It was first isolated as a constituent of protein aggregates from Alzheimer brains, distinct from the β-amyloid component[5]. Although the native function of α-synuclein is still being investigated, evidence suggests that its potential roles are in neural plasticity and the regulation of synaptic vesicle pools[6], modulation of dopamine release[7,8], alterations in dopamine synthesis[9], and targeting the dopamine transporter to the plasma membrane[10]. α-Synuclein pathology is involved in a large number of neurodegenerative diseases, including PD, dementia with LB, Lewy body variant of Alzheimer’s disease, neurodegeneration with brain iron accumulation type-1, and multiple system atrophy, which is mostly due to the remarkable discovery in dominantly inherited α-synuclein substitutions. Missense mutations (A53T, A30P, and E46K) in the α-synuclein gene were found to be the cause of autosomal dominant PD in a small group of Mediterranean[11], German[12], and Spanish families[13]. Among them, A53T was found in at least 12 families with familial PD, although these families likely to share a common ancestor[14,15] and A53T seems to induce more severe harm either in vivo or in vitro than A30P. The pathological consequences of the A53T α-synuclein mutation is closely associated with PD, whose symptoms include reduced spontaneous movement, static tremor, muscular rigidity, progressive inability to maintain erect posture, and shortening of the step length[16], and neurons in the substantia nigra are predominantly, but not exclusively affected[17].
1-Methyl-4-phenylpyridinium (MPP+), the active metabolite of 1-methyl-4-phenyl-2,3,6-tetrahydropyridine, can enter the mitochondria to induce oxidative stress and impair energy metabolism[18] by inhibiting mitochondrial complex I[19,20], inducing a syndrome closely resembling PD. The neurotoxicity of exogenous dopamine (DA) has been described in in vivo primary cultures and several cell lines[21,22]. In dopaminergic neurons, DA is oxidized easily in vitro and in vivo to a variety of neurotoxic metabolites such as highly cytotoxic quinine molecules[23,24], which participate in the generation of reactive oxygen species. Many studies indicate that an imbalance between cytoplasmic and vesicular DA may lead to oxidative stress and cell degeneration[25]. DA may cause apoptosis, but most studies on DA toxicity agree that non-apoptotic mechanisms are also involved in DA-induced cell death[26,27].
Because a slowly progressive brain disorder is difficult to study in humans, a simple, reliable, and valid method is needed for furthering our understanding of the role of α-synuclein in PD and for screening compounds with therapeutic potential. SH-SY5Y neuroblastoma is a widely and extensively used target cell line in the assessment of neurotoxicity and neuroprotection. So we sought to create a cell-based model by overexpressing mutant A53T human α-synuclein in SH-SY5Y cells.
Materials and methods
Cell culture Human dopaminergic neuroblastoma cells SH-SY5Y (American Type Culture Collection, ATCC, Manassas, VA, USA) were maintained at 37 °C in 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, Grand Island, NY, USA), supplemented with 10% fetal bovine serum (Hyclone, Logan, UT, USA), 100 U/mL penicillin, and 0.1 g/L streptomycin (Gibco, USA).
Plasmid construction Total RNA was extracted from cultured cells by the TRIzol (Invitrogen, Carlsbad, CA, USA) extraction method. From 2 µg of total RNA, cDNA was synthesized using 200 units of reverse transcriptase (Super-scriptTM III RT, Invitrogen, USA) and oligo (dT) primers in a final volume of 20 µL. The following primer pairs were used when processing PCR with pfuUltra (Stratagen, La Jolla, CA, USA): primers α-synuclein-F (5'-GAA CTC GAG GGA CTC AGT GTG GTG-3' with the Xho I site) and α-synuclein-R (5'-CT TCT AGA GGA TGG AAC ATC TGT CAG C-3' with the Xbal I site) for α-synuclein (524 bp), and GAPDH-F (5'-CTC ATG ACC ACA GTC CAT GC-3') and GAPDH-R (5'-CAC CAC CCT GTT GCT GTA GC-3') for glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 456 bp). PCR amplification was conducted under the following conditions: for α-synuclein, initial denaturation was at 94 °C for 2 min, 30 s at 94 °C, 30 s at 57 °C, and 50 s at 72 °C for 30 cycles, followed by 10 min at 72 °C. For GAPDH, initial denaturation was at 94 °C for 2 min, 30 s at 94 °C, 30 s at 59 °C, and 30 s at 72 °C for 30 cycles, followed by 10 min at 72 °C. After amplification, 50 µL aliquots were electrophoresed in 1% agarose gel (Biowest, Miami, FL, USA), followed by photographic recording of the gel stained with ethidium bromide. Then, the product of the PCR was obtained by agarose gel DNA purification (TaKaRa, Tokyo, Japan) and ligated into the pMD18-T simple vector (TaKaRa, Japan) after adding DeoxyAdenosine Triphosphate (dATP) to the blunt terminal of the fragment. The positive transformants were screened by bacteria PCR and DNA sequencing after transforming to competent Escheria coli TG-1. The A53T mutant, human α-synuclein gene was procured by gene splicing and overlap extension (SOE) on site-directed mutagenesis of wild-type human α-synuclein which was inserted into a pMD18-T simple vector purified by PureLinkTM Hipure plasmid DNA purification kits (Invitrogen, USA). The following primers were used in the SOE: forward (5'-TGC ATG GTG TGA CAA CAG TGG CTG AGA-3'), reverse (5'-GCC ACT GTT GTC ACA CCA TGC ACC ACT C-3'). The A53T, mutant human α-synuclein cDNA was sub-cloned into the pcDNA3.1(+) vector (Invitrogen, USA) after double digestion of the Xho I and Xbal I restriction enzymes to either insert DNA or the vector.
Transfection and selection One day before transfection, 2×105 cells were seeded in 500 µL of growth medium without antibiotics in a 24-well format so that cells would be 90%–95% confluent at the time of transfection. DNA was diluted in 50 µL Opti-MEM I Reduced Serum Medium (Invitrogen, USA) without serum, and the appropriate amount of Lipofectamine™ 2000 (Invitrogen, USA) was diluted in 50 µL of Opti-MEM I Medium. After incubation for 5 min at room temperature, the diluted DNA was combined with the diluted Lipofectamine™ 2000, mixed gently, and incubated for 20 min at room temperature. We added 100 µL complexes to each well containing the cells and medium. The cells were incubated at 37 °C in a CO2 incubator for 18–48 h. During incubation, the medium was changed after 4–6 h, and then cells were passaged at 1:10 into fresh growth medium after incubation. The cells were incubated in the medium containing 0.6g/L G418 (Gibco, USA) for 14 d to select the stably-transfected cells, and then the cells were collected for monoclone screening. 100 µL medium containing 0.3 g/L G418 was added to all of the wells in the 96-well plate except well Al which was left empty. 200 µL cell suspension was added to well A1, then 100 µL cell suspension was quickly transferred from the first well to well B1 by gently pipetting and repeating the same procedure as H1. 100 µL of cell suspension was discarded from H1 so that it ended up with the same volume as the wells above it. An additional l00 µL medium was added to each well in column 1 (A1–H1), and then 100 µL cell suspension was quickly transferred from the wells in column 1 to those in column 2 (A2–H2) with gently pipetting and repeating the same procedure as that of column 12. Cell suspension l00 µL was discarded from each well in the last column (A12–H12) so that all of the wells ended up with 100 µL of cell suspension. The final volume of all the wells was brought to 200 µL by adding 100 µL medium to each well. Each well that contained just a single cell was checked and marked so that these monoclones could be subcultured from the wells into larger vessels. We did the subsequent work listed below after the cells were cultured in DMEM medium containing 0.3 g/L G418 for about 3 months.
Western blotting Protein concentration was determined by the Bradford assay (Applygen, Beijing, China). Equal amounts of protein were loaded onto each lane, and electrophoresed on SDS-PAGE with Tris-glycine running buffer. They were then transferred to nitrocellulose membranes by wet electrotransfer for 90 min at 100 mA. The blocked membranes were incubated overnight at 4 °C with the polyclonal antihuman α-synuclein antibody (R&D, Minneapolis, USA 1:1000 dilution). Following 1 h of incubation at room temperature with a horseradish-peroxidase (HRP)-coupled secondary antibody (1: 5000 dilution), the blots were washed and immunodetection was carried out using enhanced chemiluminiscence detection reagents (Amersham Bio-sciences, Piscataway, NJ, USA). The blots were then stripped and reprobed with the monoclonal antibody against β-actin (Sigma, St Louis, CA, USA, 1:1000 dilution) and detected as described earlier.
Immunocytochemistry After fixation with 4% paraformal-dehyde, the cell slides were rinsed in phosphate buffer solution (PBS) and pre-incubated with 3% H2O2 for 10 min, and permeabilized with 0.5% Triton X-100 in PBS for 5 min. Then, the cells were blocked with normal rabbit serum for 15 min at room temperature. The following primary antibody (R&D, USA, 1:100 dilution) was then added for overnight incubation at 4 °C in a humidified chamber. After rinsing for 3×3 min in PBS, the cells were incubated with secondary biotinylated antibody at 37 °C for 15 min, followed by rinsing for 3×3 min in PBS. Then, the cells were incubated with HRP-labeling streptavidin/avidin working solution at 37 °C for15 min. The final reaction product was visualized with chromogen diaminobenzidine (DAB; Sigma, USA).
RT-PCR A total RNA of 2 µg from the transfected and non-transfected cells as mixed with the RT reaction mixture (Invitrogen, USA). After reverse transcription, Platinum Taq DNA polymerase (Invitrogen, USA) and the samples (total volume 50 µL) were placed in a thermal cycler. The following primer pairs were used when processing PCR: primers α-synuclein-F (5'-GAA GCA GAG GGA CTC AGT GTG GTG-3') and α-synuclein-R (5'-CT TGT ACA GGA TGG AAC ATC TGT CAG C-3') for α-synuclein (524 bp), β-actin-F (5'-CCT CGC CTT TGC CGA TCC-3'), and β-actin-R (5'-GGA TCT TCA TGA GGT AGT CAG TC-3') for β-actin (620 bp). PCR amplification of α-synuclein was conducted under the same conditions listed above in plasmid construction, and the amplification of β-actin was conducted under the following conditions: initial denaturation at 94 °C for 2 min, 30 s at 94 °C, 30 s at 55 °C, and 60 s at 72 °C for 30 cycles, followed by 10 min at 72 °C. After PCR, 10 µL aliquots of the reaction mixtures were resolved on 1% agarose gel containing ethidium bromide to identify the DNA amplicons generated.
Cell viability MTT assay is a standard method used to assess cell viability. Briefly, the cells (5×103 cells/well) were seeded in 96-well plates. After exposure to various concentrations (100, 200, and 500 µmol/L) of MPP+ and DA for 24, 48, and 72 h, 10 µL MTT, (5 g/L in PBS, Sigma, USA) solution was added to each well and the plates were incubated for an additional 4 h at 37 °C. Then, the MTT solution in medium was aspirated off. To achieve solubilization of the formazan crystals formed in the viable cells, 200 µL DMSO was added to each well. The absorbance was read at 570 nm with DMSO as the blank.
Flow cytometry (Fluorescence-activated cell sorter analysis) To detect early apoptosis and late apoptosis/necrosis, the cells were stained with fluorescein isothio-cyanate-conjugated Annexin V and propidium iodide (PI, BD Clontech San Jose, CA, USA) after being treated with 100 µmol/L MPP+ for 24 h. Approximately 1×106 cells were washed with cold PBS before being resuspended in 200 µL cold 1×binding buffer. 10 µL Annexin V-FITC and 5 µL PI were added and incubated for 15 min at room temperature in the dark. A further 300 µL binding buffer was added to terminate the reaction, and flow cytometric analysis was conducted immediately with 20 000 cells. The experiment was repeated 3 times and the results were averaged.
DNA fragmentation assay The cells were grown to about 80% confluence and then treated with 200 µmol/L MPP+ for 48 h. After treatment, the cells were resuspended in 100 µL DNA lysis buffer (TaKaRa, Japan), and then the supernatant was moved to another tube. The above step was repeated, and 200 µL of the final supernatant was collected. The liquid was incubated at 56 °C for 1 h by adding 20 µL 10% SDS and 20 µL proteinase K, and then reacted at 37 °C for 1 h after adding 20 µL RNase to allow complete RNA digestion. The DNA was precipitated with 950 µL 100% ethanol and 130 µL precipitant for 2 h at -70 °C. The DNA was pelleted at 12 000×g for 15 min and washed twice with 80% ethanol. The DNA was dissolved in Tris-EDTA buffer, and analyzed on 1.5% agarose gel for electrophoresis. DNA bands were visualized with ethidium bromide under ultraviolet light and photographed.
Compound screening The procedures were done as the part of cell viability, but the cytotoxin was 50 µmol/L DA for 24 h of incubation. Ninety-nine compounds were added to the medium at the concentration of 1×10-5 mol/L, respectively. These compounds were extracted from Fraxinus sielboldiana blume, belonging to the Oleaceae family, which is widely distributed in the east of Asia, especially in the south of China.
Statistical analysis All results were expressed as mean±SD. Statistical analysis was performed between the 2 groups by Student’s t-test. P<0.05 indicated significant difference.
Results
Construction of the eukaryotic expression system of mutant A53T human α-synuclein We obtained the mutation using the SOE method and then identified it by DNA sequencing blasted to GeneBank N
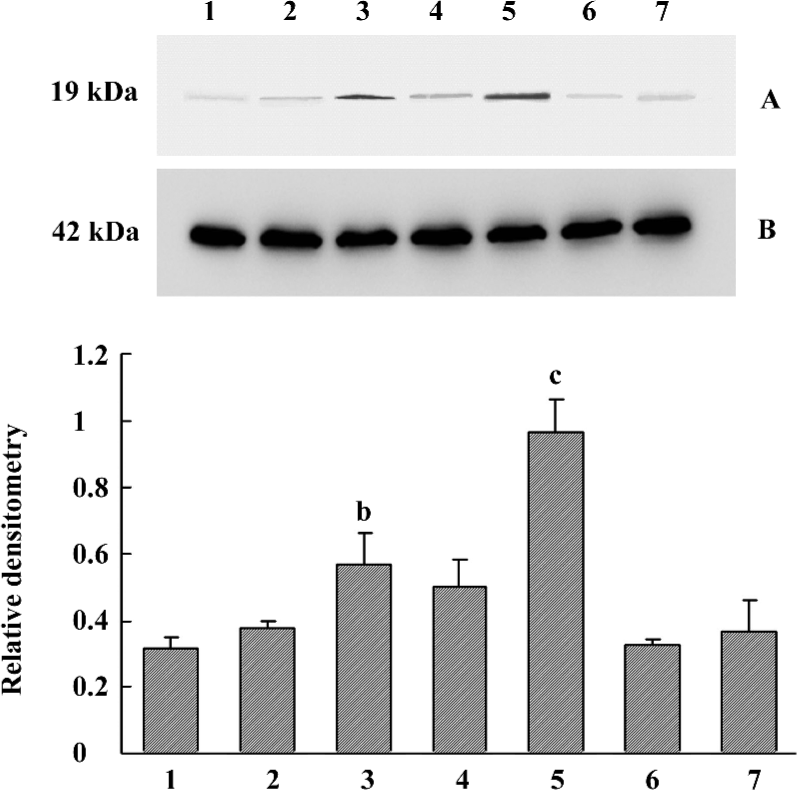
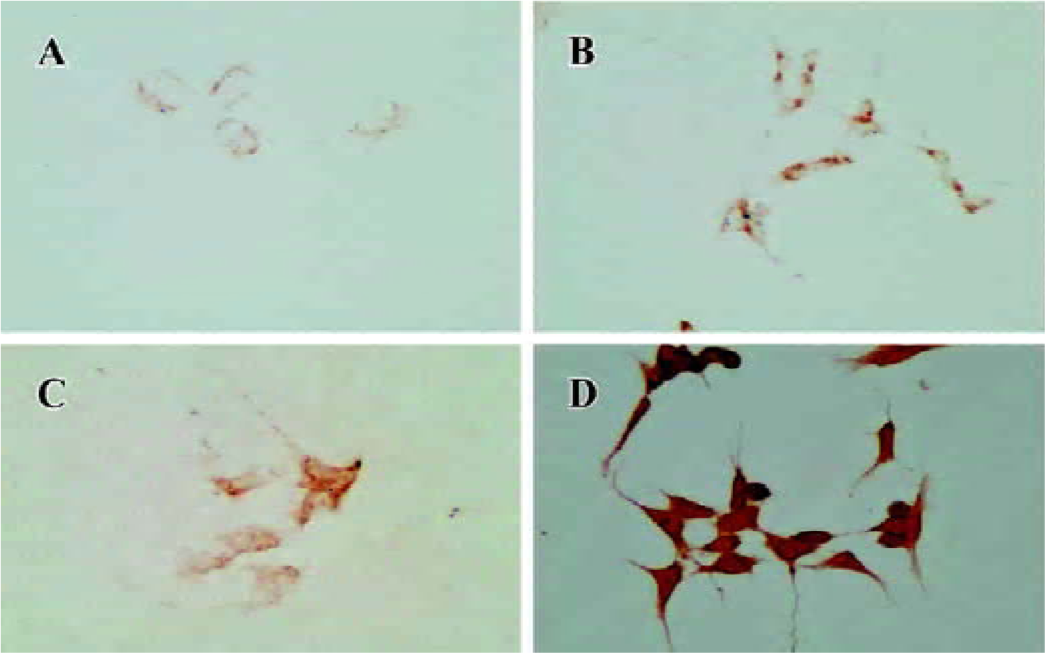
Increased α-synuclein protein expression in the cell culture model It was identified that α-synuclein expression increased in human SH-SY5Y cells after 90 d of stable transfection by Western blotting and immunocytochemistry. In the normal control or pcDNA3.1(+)-alone transfected group, the protein expression was maintained at a normal level. How-ever, transfected monoclones showed different levels (Figure 1). Through analysis by Gel-Pro Analyzer software 3.1 (Media Cybernetics, LR, USA), we found that clone 1 (P<0.05) and clone 3 (P<0.01) were significantly different compared with the control, so clone 3 was used as a cell model for our study due to the high level expression of exogenous mutant α-synuclein. The other 3 transfected groups seemed to only express the exogenous resistance gene designed in the pcDNA3.1(+) vector for their survival in the culture medium containing G418. Different clones showed different expression levels, which suggested that random integration of exogenous gene into genome was not the same copies. We also used immunocytochemistry in cell slides to characterize the cellular localization and expression of α-synuclein. The results showed that positive immunostaining of overexpression was widely localized in the cytoplasm and synapse compared with the control. Almost no staining was detected if the primary antibody was omitted (Figure 2).
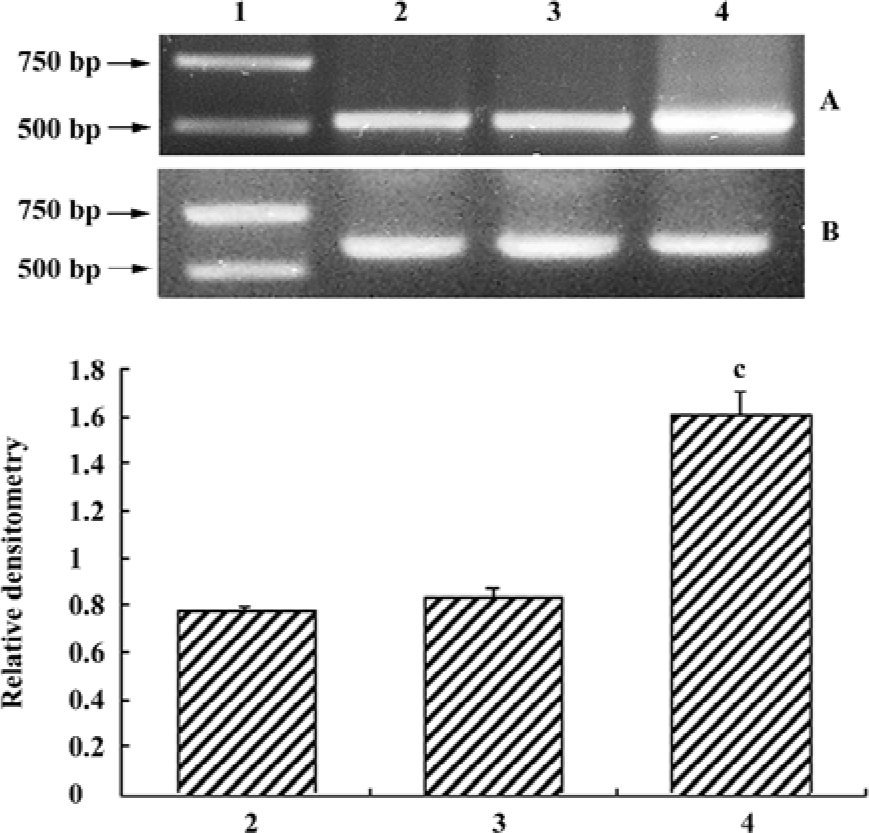
Increased α-synuclein transcriptional level in the cell model Given the change of protein expression, we sought to validate the transcriptional level obtained by semiquanti-tative RT-PCR. Clearly, α-synuclein was upregulated in the transfected cells (Figure 3).
Comparison of MPP+ and DA-induced cytotoxicity between normal and stably transfected cells All groups of cells were treated with a range of concentrations (100, 200, and 500 µmol/L) of MPP+ and DA for different times (24, 48, and 72 h), and cell viability was determined by MTT assay. MPP+ and DA induced an evident dosage and time-dependent loss in cell viability. The treatment of cells with 100, 200, and 500 µmol/L MPP+, resulted in a cell viability loss of about 2.6%, 8.1%, and 11.7% for 24 h; 20.2%, 24.6%, and 28.9% for 48 h; and 40.3%, 49.2%, and 62.8% for 72 h in the pcDNA3.1(+)-alone transfected group. However, cell viability loss was about 9.3%, 14.8%, and 25.1% for 24 h; 27.3%, 29.4%, and 45.3% for 48 h; and 49.5%, 53.8%, and 75.1% for 72 h in the clone 3 group, respectively (Figure 4A–4C). The incubation of cells with 100, 200, and 500 µmol/L DA resulted in a cell viability loss of about 56.1%, 57.6%, and 61.8% for 24 h; 69.7%, 77.1%, and 78.5% for 48 h; and 82.8%, 89.9%, and 95.3% for 72 h in the pcDNA3.1(+)-alone transfected group, while there was a loss of about 65.2%, 70.3%, and 75.8% for 24 h; 77.6%, 78.9%, and 84.2% for 48 h; and 90.0%, 94.0%, and 98.0% for 72 h in the clone 3 group, respectively (Figure 4D–4F). It was found that more rapid loss of cell proliferation occurred with the incubation of DA than MPP+, and we concluded that there was significant difference between the pcDNA3.1(+) group and the clone 3 group in cell viability when treated with MPP+ and DA. However, there was no statistical difference between normal cells and transfected cells without MPP+ or DA, and it was indicated that the cell model would not show the more severe cytotoxicity unless it was exposed to an exogenous cytotoxin.

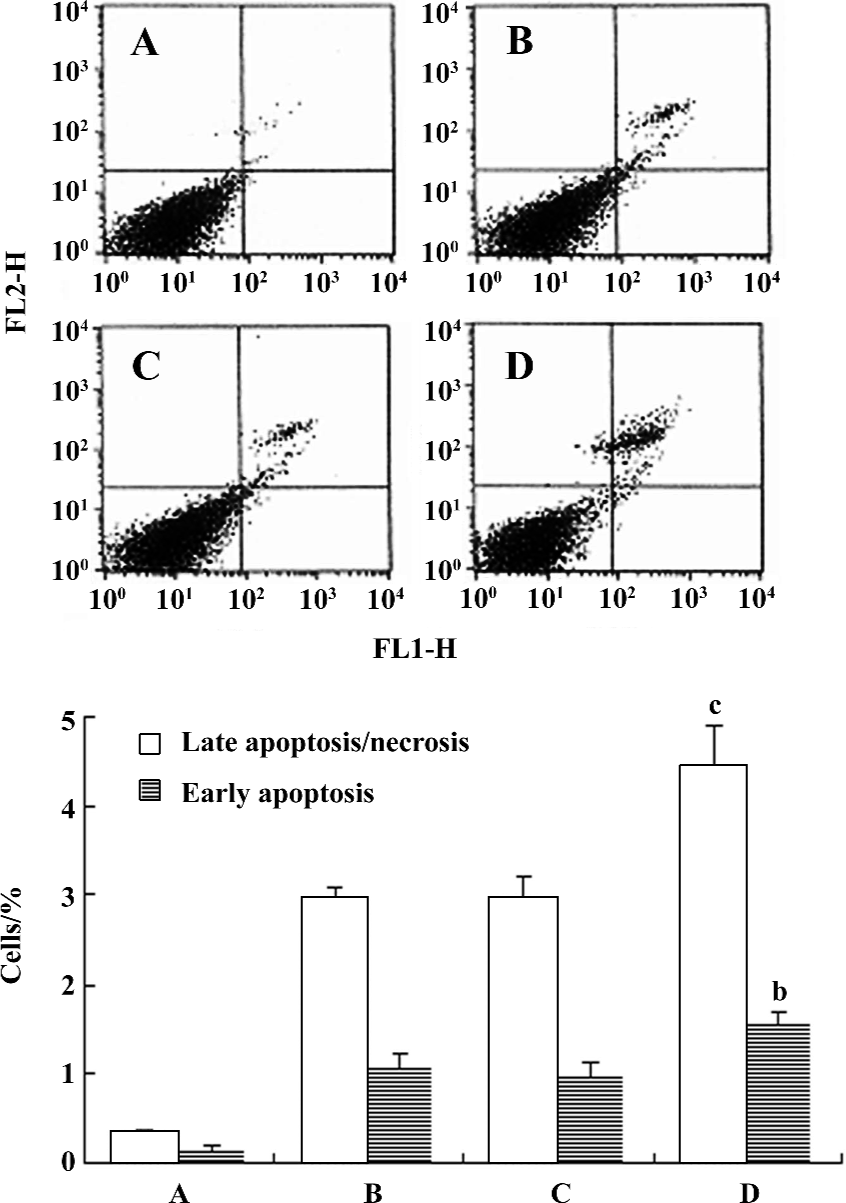
We identified early apoptotic cells, late apoptotic/necrotic cells, and viable cells by double staining the cells with PI and Annexin V. There was a significant difference between the pcDNA3.1(+)-alone transfected group and the clone 3 group after the cells were treated with 100 µmol/L MPP+ for 24 h. The percentage of both Annexin V+/PI– cells and Annexin V+/ PI+ cells increased from 0.98% to 1.56% and from 2.98% to 4.45%, respectively (Figure 5).
MPP+ has been described as an effective inducer for cell apoptosis, and a laddered pattern of DNA degradation is considered a molecular hallmark of apoptosis. Here, we proceeded to study the DNA from cells treated with 200 µmol/L MPP+ for 48 h by means of electrophoresis in agarose gels (Figure 6). In the transfected cells, an apoptotic pattern of DNA degradation was easily and reproducibly detected. However, this apoptotic pattern could not be detected in non-transfected cells.
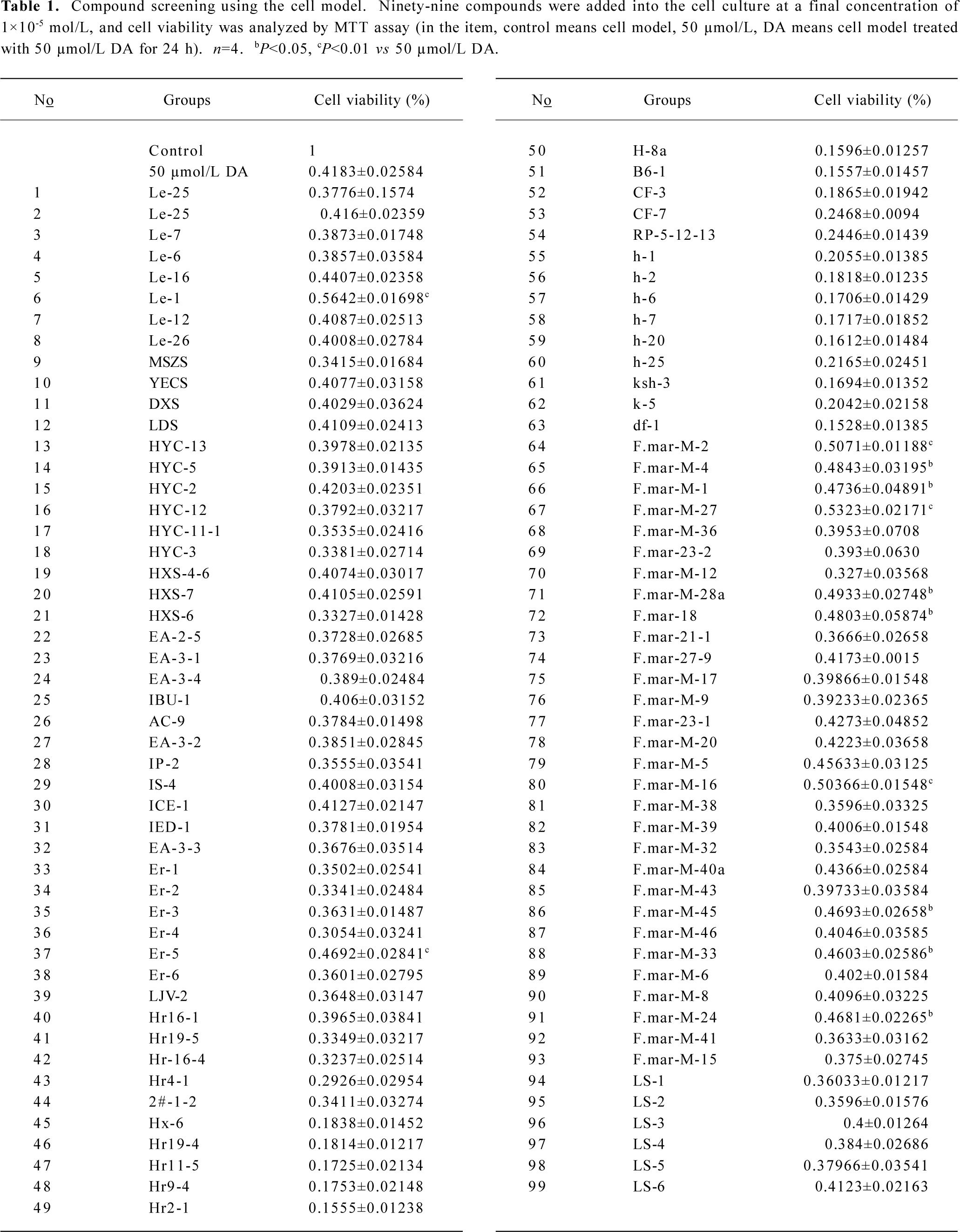
Compounds screening In the primary screening experiment, 12 of the 99 compounds showed anticytotoxic activity at the concentration of 1×10-5 mol/L (Table 1). After further screening, we found N
Full table
Discussion
In the search for a better approach to compound screens for PD, which on the one hand targets a specifically related protein, and on the other, demonstrates anticytotoxic effects against the cytotoxin. In this study, we constructed a cell-based model of α-synucleinopathy to successfully develop a SH-SY5Y cell line expressing corresponding A53T mutant α-synuclein, which could maintain the expression level continually. In other studies[28–30], several kinds of models were established for the research of α-synuclein, but not for compound screens and drug development. With the advantage of persistent expression, the cell model was suitable for long-time research work without repeated transfection.
Overexpression or aggregates of α-synuclein are found in a number of neurodegenerative disorders, now termed synucleinopathies[31]. Despite the physiological localization of α-synuclein in presynaptic terminals, the aggregates are present throughout the cell body. In our study, similarly altered localization of α-synuclein was widespread in the cell model which expressed the A53T mutant version.
Effort in developing a therapy against neurodegeneration is therefore aimed at the inhibition of overexpression or aggregate formation[32,33], but the role of proteins in the pathogenesis of human neurodegenerative disease has become an issue of debate[34] because a number of studies have not supported their toxicity[35,36]. It was demonstrated that overexpression of wild-type human α-synuclein rescues mesencephalic dopaminergic cells from MPP+-induced apoptotic cell death by attenuating cytochrome c release, caspase-3 activation, and proteolytic activation of Protein Kinase C-δ (PKC-δ). However, overexpression of the A53T human α-synuclein mutant exacerbates MPP+-induced apoptosis by augmenting proteolytic cleavage of PKCδ[37]. So by using this cell model, we can perform the studies from elementary compound screens to mechanism research of the selected compounds for drug development as this model either imitates the reaction of human neurons in vitro or simultaneously represents the interactions between overexpression of mutant α-synuclein and active compounds.
Although it has been suggested that α-synuclein aggregation may stimulate apoptosis, it remains elusive whether programmed cell death itself is directly responsible for neuronal loss, especially considering the observations that α-synuclein may also exert anti-apoptotic activity[38,39]. So its potential cytotoxic effects when encountering cytotoxin were tested in this study between normal and stably-transfected cells with MPP+ and DA on cell viability. The addition of MPP+ or DA led to more severe α-synuclein-dependent growth inhibition in transfected cells than in non-transfected cells. It was suggested that the model overexpressing mutant α-synuclein could show much more vulnerability to cytotoxin compared with normal cells. When compound screens were carried out, the candidates with anticytotoxic activity would be screened out by the cell model.
Apoptosis and necrosis are 2 modes of cell death in nucleated eukaryotic cells. Apoptosis is a programmed cell death characterized by changes in condensation of nuclear chromatin, cytoplasmic blebbing, and exposure of phosphatidylserine (PS) residues on the outside of the plasma membrane[40]. Necrosis, on the other hand, is accidental cell death and indicates the features of mitochondrial swelling, rupture of the plasma membrane, and the release of cytoplasmic constituents[41]. A key event of apoptosis in the early stage is that PS of the inner
Leaflet of the cell membrane appears in the outer leaflet, becoming a molecular marker for phagocytosis. Annexin V, which preferentially binds to PS, is very useful in detecting the expression of PS on the surface of apoptotic cells. Although neuronal apoptosis is a normal event during development, many neurodegenerative diseases are thought to involve abnormal cell death that leads to the damage of the nervous system. In our study, when MPP+ existed, there was a significant difference between the control and the transfected group, reflecting the increase of early apoptosis and late apoptosis/necrosis in addition to the appearance of the DNA “ladder”. It was concluded that the cell model showed much more sensitivity to cytotoxin than non-transfected cells, which could demonstrated the validity of the cell model.
After high throughput screening of 99 compounds, we found that 12 could decrease DA-induced cytotoxicity, and 3 of the 12 had definite anticytotoxic activity. The preliminary result indicated that the 3 compounds deserved further research because of their therapeutic potential.
In summary, this study presented a cell-based model that recapitulated pathological properties of mutant α-synuclein, which is particularly associated with the severer phenotype of familial PD[42], and demonstrated the potential cytotoxicity of A53T mutant α-synuclein. This cellular model can be used for further, fundamental studies associated with the underlying mechanisms of overexpression and genetic or environmental effects of α-synuclein-mediated cytotoxicity. The model also can be applied to the screening of drugs with therapeutic potential for synucleinopathies.
References
- Rijk MC, Tzourio C, Breteler MM, Dartigues JF, Amaducci L, Lopez-Pousa S, et al. Prevalence of Parkinsonism and Parkinson’s disease in Europe: the europarkinson collaborative study. European community concerted action on the epidemiology of Parkinson’s disease. J Neurol Neurosurg Psychiatry 1997;62:10-5.
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998;392:605-8.
- George JM, Jin H, Woods WS, Clayton DF. Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron 1995;15:361-72.
- Jakes R, Spillantini MG, Goedert M. Identification of two distinct synucleins from human brain. FEBS Lett 1994;345:27-32.
- Ueda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, . Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci USA 1993; 90: 11 282–6.
- Murphy DD, Rueter SM, Trojanowski JQ, Lee VMY. Synucleins are developmentally expressed, and α-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J Neurosci 2000;20:3214-20.
- Abeliovich A, Schmitz Y, Farinas I, Choi Lundberg D, Ho WH, Castillo PE, et al. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 2000;25:239-52.
- Stefanis L, Larsen KE, Rideout HJ, Sulzer D, Greene LA. Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J Neurosci 2001;21:9549-60.
- Perez RG, Waymire JC, Lin E, Liu JJ, Guo F, Zigmond MJ. A role for alpha-synuclein in the regulation of dopamine biosynthesis. J Neurosci 2002;22:3090-9.
- Lee FJ, Liu F, Pristupa ZB, Niznik HB. Direct binding and functional coupling of alpha-synuclein to the dopamine transporters accelerate dopamine-induced apoptosis. J FASEB 2001;15:916-26.
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 1997;276:2045-7.
- Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, et al. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet 1998;18:106-8.
- Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 2004;55:164-73.
- Golbe LI. α-Synuclein and Parkinson’s disease. Mov Disord 1999;14:6-9.
- Spira PJ, Sharpe DM, Halliday G, Cavanagh J, Nicholson GA. Clinical and pathological features of a Parkinsonian syndrome in a family with an Ala53Thr α-synuclein mutation. Ann Neurol 2001;49:313-9.
- Marsden CD. Slowness of movement in Parkinson’s disease. Mov Disord 1989;4:S26-37.
- Braak H, Del Tredici K, Rub U, de Vos RAI, Jansen Steur ENH, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 2003;24:197-210.
- Speciale SG. MPTP: insights into parkinsonian neurodegenera-tion. Neurotoxicol Teratol 2002;24:607.
- Beal MF. Experimental models of Parkinson’s disease. Nat Rev Neurosci 2001;2:325-34.
- Blum D, Torch S, Lambeng N, Nissou M, Benabid AL, Sadoul R, et al. Molecular pathways involved in the neurotoxicity of 6-OHDA, dopamine and MPTP: contribution to the apoptotic theory in Parkinson’s disease. Prog Neurobiol 2001;65:135-72.
- Gomez Santos C, Ferrer I, Santidrian AF, Barrachina M, Gil J, Ambrosio S. Dopamine induces autophagic cell death and α-synuclein increase in human neuroblastoma SH-SY5Y cells. J Neurosci Res 2003;73:341-50.
- Junn E, Mouradian MM. Apoptotic signaling in dopamine-induced cell death: the role of oxidative stress, p38 mitogen-activated protein kinase, cytochrome c and caspases. J Neurochem 2001;78:374-83.
- Holtz WA, O’Malley KL. Parkinsonian mimetics induce aspects of unfolded protein response in death of dopaminergic neurons. J Biol Chem 2003;278:19367-77.
- Stokes AH, Hasting TG, Vrane KE. Cytotoxic and genotoxic-potential of dopamine. J Neurosci Res 1999;55:659-65.
- Barzilai A, Melamed E, Shirvan A. Is there a rationale for neuroprotection against dopamine toxicity in Parkinson’s disease? Cell Mol Neurobiol 2001;21:215-35.
- Graeber MB, Moran LB. Mechanisms of cell death in neurodegenerative diseases: fashion, fiction, and facts. Brain Pathol 2002;12:385-39.
- Han BS, Homg HS, Choi WS, Markelonis GJ, Oh TH, Oh YJ. Caspase-dependent and -independent cell death pathways in primary cultures of mesencephalic dopaminergic neurons after neurotoxin treatment. J Neurosci 2003;23:5069-78.
- Lotharius J, Barg S, Wiekop P, Lundberg C, Raymon HK, Brundin P. Effect of mutant alpha-synuclein on dopamine homeostasis in a new human mesencephalic cell line. J Biol Chem 2002;277:38884-94.
- Lo Bianco C, Ridet JL, Schneider BL, Deglon N, Aebischer P. Alpha-synucleinopathy and selective dopaminergic neuron loss in a rat lentiviral-based model of Parkinson’s disease. Proc Natl Acad Sci USA 2002;99:10813-8.
- Yamada M, Iwatsubo T, Mizuno Y, Mochizuki H. Overexpression of alpha-synuclein in rat substantia nigra results in loss of dopaminergic neurons, phosphorylation of alpha-synuclein and activation of caspase-9: resemblance to pathogenetic changes in Parkinson’s disease. J Neurochem 2004;91:451-61.
- Wakabayashi K, Hayashi S, Kakita A, Yamada M, Toyoshima Y, Yoshimoto M, et al. Accumulation of alpha-synuclein/NACP is a cytopathological feature common to Lewy body disease and multiple system atrophy. Acta Neuropathol (Berl) 1998;96:445-52.
- Demaimay R, Chesebro B, Caughey B. Inhibition of formation of protease-resistant prion protein by Trypan Blue, Sirius Red and other Congo Red analogs. Arch Virol Suppl 2000;16:277-83.
- Heiser V, Scherzinger E, Boeddrich A, Nordhoff E, Lurz R, Schugardt N, et al. Inhibition of huntingtin fibrillogenesis by specific antibodies and small molecules: implications for Huntington’s disease therapy. Proc Natl Acad Sci USA 2000;97:6739-44.
- Sisodia SS. Nuclear inclusions in glutamine repeat disorders: are they pernicious, coincidental, or beneficial? Cell 1998;95:1-4.
- Cummings CJ, Reinstein E, Sun Y, Antalffy B, Jiang Y, Ciechanover A, et al. Mutation of the E6-AP ubiquitin ligase reduces nuclear inclusion frequency while accelerating polyglutamine-induced pathology in SCA1 mice. Neuron 1999;24:879-92.
- Huynh DP, Figueroa K, Hoang N, Pulst SM. Nuclear localization or inclusion body formation of ataxin-2 are not necessary for SCA2 pathogenesis in mouse or human. Nat Genet 2000;26:44-50.
- Kaul S, Anantharam V, Kanthasamy A, Kanthasamy AG. Wild-type α-synuclein interacts with pro-apoptotic proteins PKCδ and BAD to protect dopaminergic neuronal cells against MPP+-induced apoptotic cell death. Mol Brain Res 2005;139:137-52.
- da Costa CA, Ancolio K, Checler F. Wild-type but not Parkinson’s disease-related Ala-53-Thr mutant α-synuclein protects neuronal cells from apoptotic stimuli. J Biol Chem 2000; 275: 24 065–9.
- Kalivendi SV, Cunningham S, Kotamraju S, Joseph J, Hillard CJ, Kalyanaraman B. Alpha-synuclein up-regulation and aggregation during MPP-induced apoptosis in neuroblastoma cells: intermediacy of transferrin receptor iron and hydrogen peroxide, J Biol Chem 2004; 279: 15 240–7.
- Cohen JJ, Duke RC, Fadok VA, Sellins KS. Apoptosis and programmed cell death in immunity. Annu Rev Immunol 1992;10:267-93.
- Majno G, Joris I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am J Pathol 1995;146:3-15.
- Munoz E, Oliva R, Obach V, Marti MJ, Pastor P, Ballesta F, et al. Identification of Spanish familial Parkinson’s disease and screening for the Ala53Thr mutation of the alpha-synuclein gene in early onset patients. Neurosci Lett 1997;235:57-60.