
In vitro and in vivo evaluation of a novel oral insulin formulation1
Introduction
The lack of oral options for peptide/protein drugs limits their commercial use as well as their clinical application. To obtain an optimistic bioavailability of peptide/protein drugs by the oral route, various obstacles must be overcome, of which harsh conditions of the gastrointestinal tract and low membrane permeability are the main barriers. Enzyme inhibitors, permeation enhancers, multifunctional polymers and encapsulation technology such as microspheres and nanoparticles have been set forth to boost oral insulin bioavailability[1-5]. Recently, oil-based self-emulsifying formulations have attracted much attention as a promising oral delivery form for improving drug solubilization and dissolution and/or intestinal absorption enhancement. Both the self-emulsifying drug delivery systems (SEDDS) for lipophilic drugs and water-in-oil (W/O) microemulsion for hydrophilic molecules are within the context of self-emulsifying formulation. SEDDS are isotropic mixtures of oil, surfactant, co-surfactant and lipophilic drugs. As an outstanding successful oral formulation based on SEDDS, cyclosporine A (Sandimmun Neoral®, Basel, Basel-Stadt, Switzerland) has been developed and marketed. Generally, W/O microemulsion developed to overcome metabolic and physical barriers impermeable to water-soluble molecules, such as peptides and proteins, contains mixtures of surfactants and oil with the aqueous phase being maintained to below 20% (w/w)[6-9].
We successfully developed a novel oral insulin formulation which can form fine microemulsion with a droplet size of about 260 nm when introduced into water under mild agitation.
In this study, insulin-loaded formulation in a narrow size range was well prepared. The formulation was evaluated by various physicochemical parameters such as droplet size, insulin encapsulation efficiency and stability. The influence of this formulation on Caco-2 monolayer cells was assessed in terms of Papp and TEER. In addition, a 2-cross-over trial was conducted in diabetic beagles as an in vivo model to evaluate the potential application of this preparation in blood glucose control.
Materials and methods
Materials and animals Porcine zinc insulin (28.1 IU/mg) was purchased from Wanbang Pharmaceutical Company (Xuzhou, China). Polyethylene glycol-8-glycol octanoate/decanoate and polyglycerol-3-oleate were supplied by Cerdex (Gennee-villiers, Cerdex, France). Fetal bovine serum and trypsin-EDTA were supplied by Hyclone (Hyclone, UT, USA). Dulbecco’s modified Eagle’s media (DMEM), L-glutamine, penicillin and streptomycin were obtained from Gibco Laboratory (Invitrogen CO, Grand Island, NY, USA). Ranitidine and propranonol were purchased from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, Beijing, China). All other chemical reagents were of high purity or HPLC grade.
Male Beagle dogs (weight: 10 kg) were supplied by the large animal house of the China Academy of Military Medical Sciences (Beijing, Beijing, China). All animal experiments were compliant with local legislation “rinciples of Laboratory Animal Care”.
Formulation preparation Formulation with or without insulin was prepared according to the patient[10]. Briefly, the weighed amount of insulin was first dissolved in a few drops of 0.1 mol/L HCl in order to facilitate dissolution; NaCl solution (0.9%, w/v) was then added and the pH was adjusted to 3.0, resulting in solution A with a final insulin concentration of 60 mg/mL. Solution A was mixed with 5-fold liquid non-ionic surfactant polyethylene glycol-8-glycol octanoate/decanoate, then magnetically stirred for a few minutes at 20 °C until an optically clear formulation was observed, resulted in solution B. Solution B was then added to another liquid non-ionic surfactant polyglycerol-3-oleate, wherein the volume ratio of solution B to that of the polyglycerol-3-oleate was 1:2. The solution was mixed quickly to form a homogeneous formulation, which was stored at 4 °C. The insulin-free formulation was prepared exactly the same as the insulin-loaded formulation.
Evaluation of the formulation Droplet size: Microemulsions were formed by 1:800 dilution of the formulation with purified water. The droplet sizes of the microemulsions were determined by the photon correlation spectroscopy method using Zeta/sizer 3000HS (Malvern, Worcestershire, UK).
Encapsulation efficiency: A dialysis bag (mw cutoff: 12 000‒14 000) containing 2 mL insulin-loaded formulation and 8 mL phosphate-buffered saline buffer (PBS, pH=7.4) was stirred in 200 mL PBS buffer up to 24 h. Samples 1 mL were extracted from the PBS buffer at the time points of 0, 1, 2, 5, 10 and 24 h, and the same volume of fresh PBS was supplied to keep constant volume. The amount of insulin in the water phase was measured with Radio Immunoassay (RIA) by the method described in RIA kit instruction.
The encapsulation of insulin in the formulation was calculated according to Equation 1:
En=(Tinsulin‒insulin)/Tinsulin×100% (1)
Where En indicates encapsulation efficiency, Tinsulin (in IU/mL) is the amount of insulin added in the formulation and Winsulin (in IU/mL) is the amount of insulin in the water phase.
Physical stability: The physical stability of the formulation stored at 4 °C was evaluated by periodic visual inspection of the presence of macroscopic phase separation, as shown by cloudiness or the formation of 2 distinct layers. The pharmaceutical activity of insulin encapsulated in the formulation was measured by RP-HPLC. Before analyzing, the insulin-loaded microemulsion samples were pretreated with 0.02% Triton X-100 solution.
Proteolytic studies Native insulin (400 μg) or equimolar amounts of insulin-loaded formulation was dissolved in 920 μL (0.1 mol/L) Tris-HCl (pH 8.0); 80 μL of a 200μg /mL trypsin solution was added into the same buffer and the mixtures were then incubated at 37°C. At scheduled points (0, 5, 20, 35, 60, 90, 120, 150 and 180 min), 50 μL samples were withdrawn and diluted with the same volume of 0.1% trifluoroacetic acid to terminate the enzymatic reaction. Samples were stored at 4 °C before analysis.
Cell culture Caco-2 cell line was obtained from the American Type Culture Collection (Rockville, MD, USA). Cells were cultured as described by the supplier with DMEM in tissue culture flasks (75 cm2) in a humidified (95% relative humidity), 37 °C, 5% CO2 incubator. DMEM contained glucose (4.5 g/L) supplemented with 10% FBS, 1% NEAA, 1% L-glutamine, 100 IU/mL penicillin and 100 μg/mL streptomycin. For the transport study, cell passages between 40‒45 were used. After reaching 80% confluence in a week, cells were harvested with trypsin-EDTA and seeded in the apical sides of 12-well trans-well plates (pore size 3.0 祄, diameter 24 mm; Costar, NL, USA) at a density of 1.0×105 cells/mL. The integrity of Caco-2 monolayers was monitored by measuring the resistance across the cell layer (TEER) with an epithelial voltohmmeter (World Precision Instrument, Sarasota, Florida, USA).
Transport study After 21 d growth, the TEER across the Caco-2 monolayer cells reached an equilibrium value, indicating that the monolayer’s tight junctions had well formed and could be used for transport study[4,11]. The test drugs, insulin (50 IU/mL), ranitidine (1.5 mg/mL) and propranonol (1.5 mg/mL), were respectively dissolved in Hanks? Balanced Salt Solution (HBSS) (insulin was first dissolved in 0.01 mol/L HCl solution) and added to the different apical sides (each 0.5mL) of the chambers, Fresh HBSS was added into the basolateral side of these chambers (each 1.5 mL). In the test group, 2% (v/v) formulation was added into apical sides, while no formulation was added to the control group. Samples 200 μL were taken at given time intervals (0, 30, 60, 90, 120 and 150 min) from the basolateral sides of the chambers, and the same volume of fresh HBSS was supplied to keep constant volume in the basolateral sides. TEER values were monitored during the experiment.
The chambers were washed twice with DMEM as soon as the transport study finished. After 0.5 mL and 1.5 mL DMEM containing 10% FBS were respectively added to the apical and basolateral sides, the chambers were placed in an incubator. TEER values were measured 3 h later.
Papp (in cm/s) of drugs were calculated according to Equation 2:
Papp=(ΔQ/Δt)/(C0譇) (2)
Where ΔQ/Δt (in mmol/s) indicates linear apparent rate of mass in basolateral side, C0 (in mmol/cm3) is the initial concentration of drugs in the apical side; and A (in cm2) is the surface area of the well.
Hypoglycemic effects in diabetic beagles A 60 mg/kg dose of alloxan was intravenously injected to beagles to render diabetes. About 1 week later, the blood glucose levels were measured in 3 consecutive days in order to verify the diabetic state of the dogs. Only the animal with a glycemia level around 250 mg/dL in all 3 occasions could be considered the successful diabetic dog. Eventually, 6 diabetic dogs with similar blood glucose concentrations were used for the study. The serum glucose levels of these 6 diabetic dogs were measured (blank control) during 24 h. A week later, a 2-cross test was employed to minimize individual varieties, in which 6 diabetic dogs were randomly divided into 2 groups (each consisting of 3 dogs):, (A) oral administration of insulin-loaded formulation (2.5 IU/kg) and (B) subcutaneous administration of native insulin (0.5 IU/kg). In another week, group A were treated by the subcutaneous injection of native insulin (0.5 IU/kg), and group B were orally administered with insulin-loaded formulation (2.5 IU/kg). Blood samples were collected from the jugular vein at the given time intervals and determined using genuine test strips (Life Scan, Milpitas, CA, USA). Before the experiment, the animals were made to fast overnight, but had access to water.
The oral bioavailability (BA) was calculated by utilizing Equation 3:
BA=[AUC0-24 oral×(weight/dose)oral]/
[AUC0-24 sc×(weight/dose)sc]×100% (3)
The AUC0-24 is the area under the reduction of serum glucose levels from 0 to 24 h; weight (in kg) is the body weight of the beagles and dose (in IU) is the amount of insulin administered to the animals.
HPLC analysis The chromatography was equipped with a Waters HPLC system (Waters, Milford, Mass, USA). A 5 μL sample was injected. A VYDAC-218TP54 C18 reversed-phase column (Vydac, Hesperia, CA, USA) was used in the study.
For insulin analysis, the mobile phase consisted of 50 mmol/L NaH2PO4 (pH 3.00; A) and acetonitrile (B). The procedure was as follows: a linear gradient from 80% channel A to 20% in 20 min, channel B correspondingly changed from 20% to 80%, then returned to 80% A and 20% B within 5 min and stayed that way for 5 min before the next injection. The flow rate was maintained at 1.00 mL/min. Detection was performed at 214 nm.
Both ranitidine and propranolol were analyzed by VYDAC-218TP54 C18 column with isocratic elution at a flow rate at 1.0 mL/min. Ranitidine was analyzed at 320 nm, and the mobile phase used for analysis consisted of acetonitrile -30 mmol/L ammonium acetate (10:90, v/v, pH 3.0). Propranolol was detected at 294 nm[12] and the mobile phase was composed of acetonitrile -50 mmol/L phosphate buffer (40:60, v/v, pH 3.0).
Statistics Each set of experiments described was repeated at least 3 times. All values were expressed as mean±SD. Statistical differences were assumed to be reproducible with P<0.05.
Results
Characterization of the formulation Self-emulsifying properties of the formulations were demonstrated when the formulations were introduced into water under mild agitation. The mean droplet diameters of insulin-loaded formulation and insulin-free formulation were 264.7 nm and 257.2 nm, respectively, with 800-fold dilution.
Most of the insulin was still encapsulated by the microemulsion, even it had been dialyzed in bulk buffer over 24 h (Table 1).

Full table
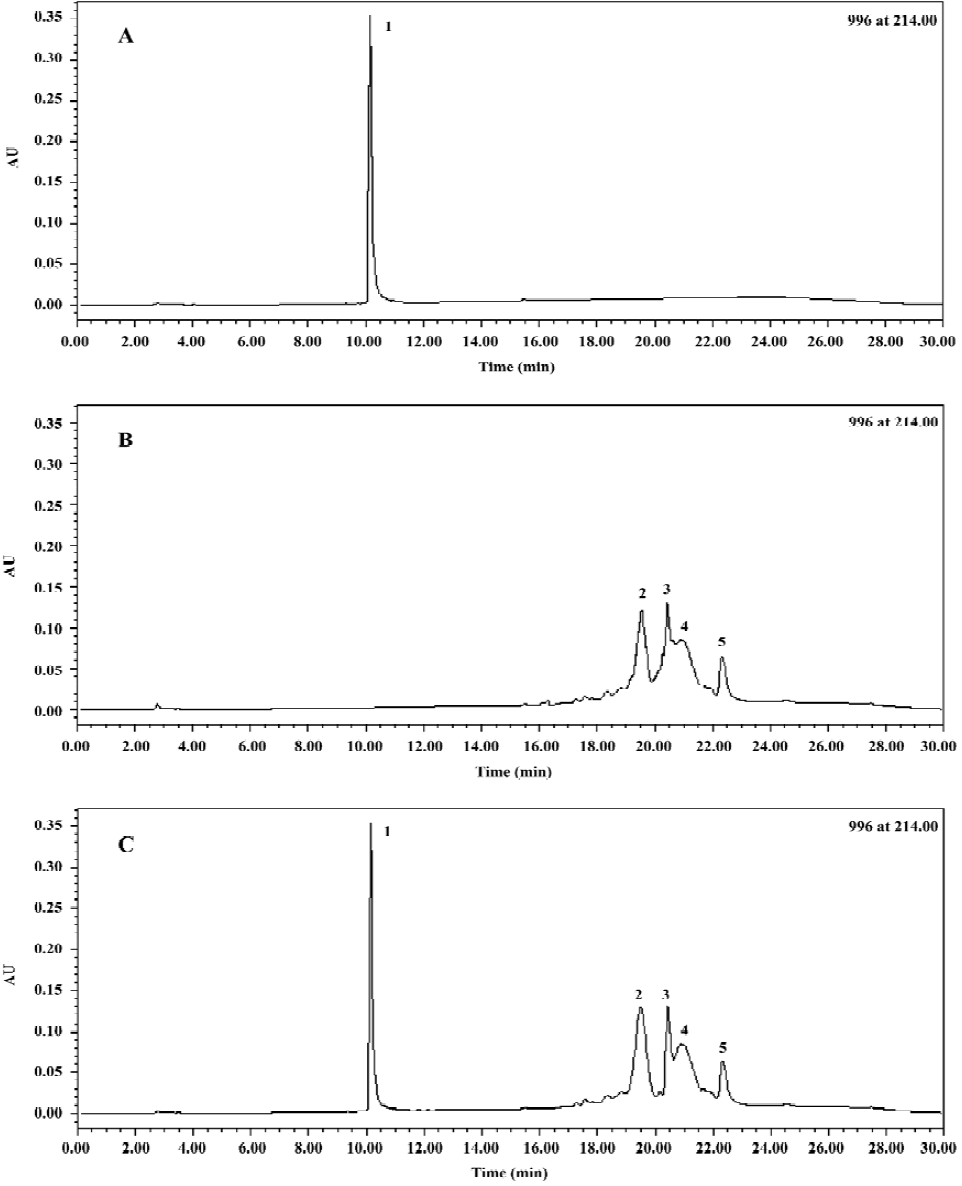
Stability study Based on our research, a single and optically clear phase was still maintained and no visible sign of change was found in both formulations during the 6-month period of storage at 4 °C. As seen in Figure 1, the HPLC chromatogram of native insulin (Figure 1A) was identical to that of the encapsulated insulin in the formulation stored for 6 months at 4 °C (Figure 1C); no other peaks appeared (regardless of surfactants; Figure 1B) compared with native insulin. Therefore, neither degradation nor conglomeration of insulin occurred in the insulin-loaded formulation.
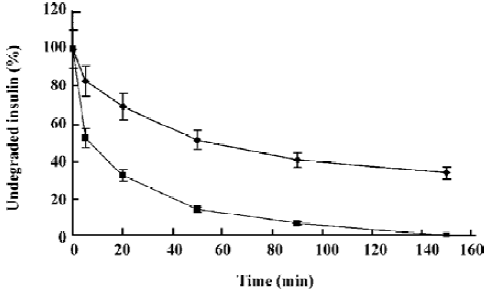
Degradation characterization The degradation profiles of native insulin and encapsulated insulin in the formulations are shown in Figure 2. More than 80% native insulin was rapidly degraded within 1 h. In contrast, the encapsulated insulin displayed a significantly higher resistance to proteolysis.
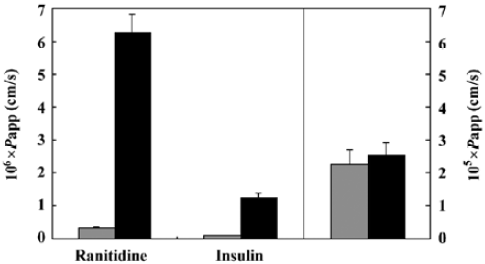
Transport study Transport studies were carried out to verify the influence of the formulation on drug transportation through the intestinal mucosa. As shown in Figure 3, the Papp of Ranitidine, a paracellular marker, increased from 0.317×10-6 to 6.26?10-6, about 20-fold higher than the control when 2% formulation was added. Insulin also showed similar behavior: its Papp increased from 0.103×10-6 to 1.23×10-6, about 12-fold higher than the control. However, the permeability of Propranolol, a transcellular marker, seemed unchanged (2.273×10-5 vs 2.533×10-5, P=0.438).
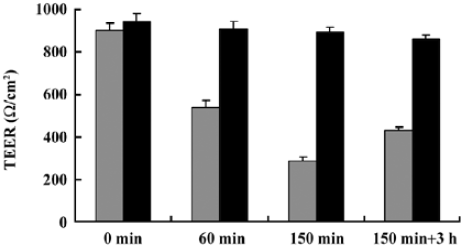
TEER studies No significant changes of TEER with time were observed in the control group. In the presence of the formulation, TEER values decreased significantly (within 150 min), but were constantly above 250 Ω/cm2, suggesting that the monolayers maintained their integrity. The effect of the formulation on Caco-2 cells appeared to be reversible as TEER values recovered (>400 Ω/cm2) after 3 h following the transportation study (Figure 4).
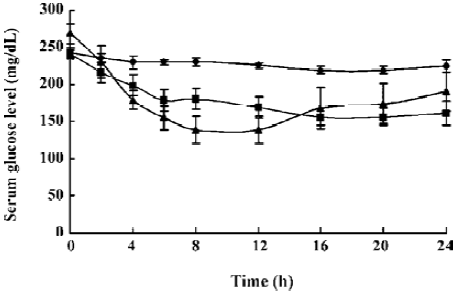
Oral efficacy of the formulation It is noteworthy that low glucose levels were maintained throughout the 24-h period. Compared with the hypoglycemic percent of native insulin (0.5 IU/kg) administered subcutaneously, the bioavailability (AUC) of this oral formulation (2.5 IU/kg) reached 15.21%. Additionally, a sustainable release was found in the diabetic dogs, which had an orally administered insulin-loaded formulation (Figure 5).
Discussion
A stable self-emulsifying formulation was developed to encapsulate insulin, which can form fine emulsions with a droplet diameter about 260 nm when introduced into water under mild agitation.
It was supposed that the ability of the formulation to encapsulate and protect insulin from proteolytic degradation may be attributed to the location of insulin, which was dissolved in the inner aqueous layer upon the effect of surfactants. Because the oil/water partition coefficient of insulin is almost 0, the diffusion of insulin across the surfactant membrane after dilution would be negligible[13]. As a result, insulin is encapsulated and protected from degradation. This structure feature makes the appropriate pretreatment necessary for a satisfactory recovery (>95%) of insulin in the formulation as determined by RP-HPLC. Without the pretreatment, the recovery of insulin in the formulation was around 30%[14].
Proteins have been reported in some studies to be absorbed at the oil/water interface and involved in the stabilization of emulsion[15,16]. The stability of our formulation was measured by visual inspection and RP-HPLC analysis, and the results showed neither physical change nor the new peak appearing in the insulin-loaded formulation even after 6 months of storage at 4 °C.
Caco-2 monolayer cells were chosen as an in vitro model to measure the ability of the formulation to enhance absorption of insulin and to explore mechanisms involved in the systemic uptake of the insulin through the gastrointestinal epithelium. Although Caco-2 cells come from the human colon carcinoma, they acquire many features of absorptive intestinal cells in culture, such as microvillous structures, hydrolysis enzymes, carrier transport systems for sugars, amino acids and drugs and well-formed tight junctions between the adjacent cells. Several reports have already shown the possibility of predicting the oral absorption of drugs in human from studies of their permeability to Caco-2 monolayers[11,17-20].
Based on the different behaviors of the 2 marker drugs, ranitidine[21] and propranolol[22], and the changes of TEER in the transport study, a safe conclusion was drawn that the tight junctions (TJ) between adjacent cells were loosened upon the influence of this formulation. So the increase of insulin transportation through Caco-2 monolayers (Papp increased from 0.103×10-6 to 1.23×10-6 with 2% formulation) may be mainly due to the changed TJ structures which allowed insulin, a hydrophilic macromolecule with 51 amino acids, to transport through epithelium membrane barriers via the transcellular route. The mechanism of the TJ “opening” was related to the C10 fatty acid, one of the main components in the formulation, which has been accepted nowadays as a substance to trigger a series of reactions leading to the widening of the TJ, thus increasing membrane permeability for hydrophilic macromolecules[23-26].
During the transport study, Caco-2 monolayers maintained integrity (TEER constantly above 250 Ω/cm2) and the TEER values recovered to 400 Ω/cm2 and above after 3 h of incubation following the transport study. Therefore, the influence of this preparation on Caco-2 cells was uninjurious and reversible with 2% formulation.
In this assay, hypoglycemia was induced by a comparative small dose of insulin (2.5 IU/kg) and the bioavailability of this oral formulation was up to 15.21% compared with sc native insulin. This posted a great cost advantage and was much more effective than the other reported studies on oral insulin delivery[1,2,27-30]. Furthermore, at a dose of 300 times more than that administered to man, no adverse effect of this preparation on the intestine, spleen and liver in mice was found, suggesting the possible use for human beings. This formulation has now been approved by the China State Food and Drug Administration to enter clinical trials.
Conclusion
A novel formulation with the capacity of encapsulating insulin was prepared. The high efficacy of blood glucose control of this formulation was attributed to such possible positive factors as degradation resistance, permeation increase and so on. Stability, efficacy and safety of the preparation make it a promising oral formulation to achieve optimistic bioavailability.
References
- Carino GP, Jacob JS, Mathiowitz E. Nanosphere based oral insulin delivery. J Control Release 2000;65:261-9.
- Whitehead K, Shen Z, Mitragotri S. Oral delivery of macromolecules using intestinal patches: applications for insulin delivery. J Control Release 2004;98:37-45.
- Pan Y, Li YJ, Zhao HY, Zheng JM, Xu H, Wei G. Bioadhesive polysaccharide in protein delivery system: chitosan nanoparticles improve the intestinal absorption of insulin in vivo. Int J Pharm 2002;249:139-47.
- Nerurkar MM, Burton PS, Borchardt RT. The use of surfactants to enhance the permeability of peptides through Caco-2 cells by inhibition of an apically polarized efflux system. Pharm Res 1996;13:528-34.
- Marschutz MK, Bernkop-Schnurch A. Oral peptide drug delivery: polymer-inhibitor conjugates protecting insulin from enzymatic degradation in vitro. Biomaterials 2000;21:1499-507.
- Kawakami K, Yoshikawa T, Hayashi T, Nishihara Y, Masuda K. Microemulsion formulation for enhanced absorption of poorly soluble drugs II. In vivo study. J Control Release 2002;81:75-82.
- New RRC, Kirby CJ. Solubilisation of hydrophilic drugs in oily formulations. Adv Drug Deliv Rev 1997;25:59-69.
- Cilek A, Celebi N, Tirnaksiz F, Tay A. A lecithin-based micro-emulsion of rh-insulin with aprotinin for oral administration: Investigation of hypoglycemic effects in non-diabetic and STZ-induced diabetic rats. Int J Pharm 2005;298:176-85.
- Gursoy RN, Benita S. Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomed Pharmacother 2004;58:173-82.
- Zheng CX, Duan MX, Ma H. Inventors; a method to prepare oily insulin formulation for oral administration. PRC Patent 1 160 122. 2004 Aug 8.
- Foss AC, Peppas NA. Investigation of the cytotoxicity and insulin transport of acrylic-based copolymer protein delivery systems in contact with Caco-2 cultures. Eur J Pharm Biopharm 2004;57:447-55.
- Modamio P, Lastra CF, Montejo O, Marino EL. Development and validation of liquid chromatography methods for the quantitation of propranolol, metoprolol, atenolol and bisoprolol: application in solution stability studies. Int J Pharm 1996;130:137-40.
- Silva-Cunha A, Grossiord JL, Puisieux F, Seiller M. W/O/W multiple emulsions of insulin containing a protease inhibitor and an absorption enhancer: preparation, characterization and determination of stability towards proteases in vitro. Int J Pharm 1997;158:79-89.
- Deng B, Liu Z, Luo G, Ma H, Duan M. Rapid quantitative determination and assessment of insulin in oil formulation by micellar electrokinetic capillary chromatography. J Pharm Biomed Anal 2002;27:73-80.
- Cournarie F, Savelli MP, Rosilio V, Bretez F, Vauthier C, Grossiord JL. Insulin-loaded W/O/W multiple emulsions: comparison of the performance of systems prepared with medium-chain-triglycerides and fish oil. Eur J Pharm Biopharm 2004;58:477-82.
- Garti N, Aserin A, Cohen Y. Mechanistic considerations on the release of electrolytes from multiple emulsions stabilized by BSA and nonionic surfactants. J Control Release 1994;29:41-51.
- Hu M, Borchardt RT. Transport of a large neutral amino acid in a human intestinal epithelial cell line (Caco-2): uptake and efflux of phenylalanine. Biochim Biophys Acta 1992;1135:233-44.
- Artursson P, Karlsson J. Correlation between oral drug absorption in humans and apparent drug permeability coefficients in human intestinal epithelial (Caco-2) cells. Biochem Biophys Res Commun 1991;175:880-5.
- Tanaka Y, Taki Y, Sakane T, Nadai T, Sezaki H, Yamashita S. Characterization of drug transport through tight junction pathway in Caco-2 monolayer: comparison with isolated rat jejunum and colon. Pharm Res 1995;12:523-8.
- Gan LSL, Thakker DR. Applications of the Caco-2 model in the design and development of orally active drugs: elucidation of biochemical and physical barriers posed by the intestinal epithelium. Adv Drug Deliv Rev 1997;23:77-98.
- Lee K, Ng C, Brouwer KL, Thakker DR. Secretory transport of ranitidine and famotidine across Caco-2 cell monolayers. J Pharmacol Exp Ther 2002;303:574-80.
- Markowska M, Oberle R, Juzwin S, Hsu CP, Gryszkiewicz M, Streeter AJ. Optimizing Caco-2 cell monolayers to increase throughput in drug intestinal absorption analysis. J Pharmacol Toxicol Methods 2001;46:51-5.
- Hayashi M, Tomita M, Awazu S. Transcellular and paracellular contribution to transport processes in the colorectal route. Adv Drug Deliv Rev 1997;28:191-204.
- Sha X, Yan G, Wu Y, Li J, Fang X. Effect of self-microemulsifying drug delivery systems containing Labrasol on tight junctions in Caco-2 cells. Eur J Pharm Sci 2005;24:477-86.
- Eaimtrakarn S, Rama Prasad YV, Ohno T, Konishi T, Yoshikawa Y, Shibata N. Absorption enhancing effect of labrasol on the intestinal absorption of insulin in rats. J Drug Target 2002;10:255-60.
- Mori S, Matsuura A, Rama Prasad YV, Takada K. Studies on the intestinal absorption of low molecular weight heparin using saturated fatty acids and their derivatives as an absorption enhancer in rats. Biol Pharm Bull 2004;27:418-21.
- Fasano A, Uzzau S. Moldulation of intestinal tight junctions by Zonula occludens toxin permits enteral administration of insulin and other macromolecules in an animal model. J Clin Invest 1997;99:1158-64.
- Cernea S, Kidron M, Wohlgelernter J, Modi P, Raz I. Comparison of pharmacokinetic and pharmacodynamic properties of single-dose oral insulin spray and subcutaneous insulin injection in healthy subjects using the euglycemic clamp technique. Clin Ther 2004;26:2084-91.
- Katsuma M, Watanabe S, Kawai H, Takemura S, Sako K. Effects of absorption promoters on insulin absorption through colon-targeted delivery. Int J Pharm 2006;307:156-62.
- Morishita M, Goto T, Nakamura K, Lowman AM, Takayama K, Peppas NA. Novel oral insulin delivery systems based on complexation polymer hydrogels: single and multiple administration studies in type 1 and 2 diabetic rats. J Control Release 2006;110:587-94.