
Effects of sodium ferulate on amyloid-beta-induced MKK3/MKK6-p38 MAPK-Hsp27 signal pathway and apoptosis in rat hippocampus
Introduction
One of the neuropathological hallmarks of Alzheimer’s disease (AD) is an accumulation of plaques consisting predominately of amyloid-beta (Aβ) peptide, which is a cleavage product of amyloid precursor protein (APP) resulting from the action of β- and γ-secretase[1]. Accompanying the accumulation of Aβ is elevation in the inflammation-related proteins of the complement cascade, as well as interleukin (IL)-1β and tumor necrosis factors-α[2]. Any or all of the above mentioned proteins are potential triggers for the neuronal death and synaptic loss. Numerous studies have shown that Aβ-induced neuronal death demonstrates signs of apoptosis[3,4]. However, the signal transduction mechanism(s) by which these losses occur remains largely unknown.
The mammalian mitogen-activated protein kinases (MAPK) can be subdivided into the extracellular signal-regulated kinases (ERKs), the Jun N-terminal kinases (JNKs), and the p38 MAPK. JNK and p38 MAPK are also called stress activated protein kinases (SAPK). These pathways appear to be activated by a wide variety of cellular stresses including heat shock, lipopolysaccharides (LPS), and inflammatory cytokinases. It has also been demonstrated that the p38 MAPK pathway is hyperactive in the AD brain[5]. Activation of p38 MAPK is mediated by the upstream MAPK kinase, referred to as mitogen-activated protein kinase kinase (MKK)3 and MKK6[6,7]. In addition, there is the potential for crosstalk between JNK and p38 MAPK pathways because of MKK4 (SEK1), which has been shown to activate both p38 MAPK and JNK[6]. Activated p38 MAPK phosphorylates MAPK activating protein kinase 2 (MAPKAPK-2), which phosphorylates the 27 kDa heat shock protein (Hsp27)[8] and activating transcription factors 2 (ATF-2)[9]. Hsp27 is a molecular chaperone with an ability to interact with a large number of proteins. Recent evidence has shown that Hsp27 regulates apoptosis through an ability to interact with key components of the apoptotic signaling pathway, in particular, those involved in caspase activation and apoptosis[10].
Sodium ferulate (SF), extracted from a traditional Chinese herbal medicine, has potent antioxidant[11] and anti-inflammatory activities[12]. It has recently been reported that long-term administration of ferulic acid protected mice against learning and memory deficits induced by centrally administered β-amyloid[13]. The primary site of action of ferulic acid could be the microglia[14] and astrocytes[15]. A recent report showed that ferulic acid inhibited the formation of Aβ fibrils and destabilized preformed fibrillary Aβ[16]. Sultana et al reported that ferulic acid ethyl ester significantly inhibited Aβ1-42-induced cytoxicity, intracellular reactive oxygen species accumulation, lipid peroxidation, and the induction of inducible nitric oxide synthase in primary hippocampal cultures[17]. In addition, ferulic acid attenuates iron-induced oxidative damage and apoptosis in cultured neurons[18] and reduces the expression of inducible nitric oxide synthase (iNOS) and cyclooxygenase activity following exposure to LPS[19]. Our previous report showed that SF had inhibitory effects on Aβ-induced p38 MAPK phosphorylation and neuronal apoptotic deaths in the rat hippocampus[20]. Therefore, in this study, we investigated the effect of SF on Aβ1-40-induced phosphorylation levels of the MKK/MKK6 and p38 MAPK as well as MAPKAPK2 and Hsp27 in rat hippocampus. We have also studied the effect of the selective p38 MAPK inhibitor SB203580 on these kinase phosphorylations and the pro-apoptotic pathways.
Materials and methods
Materials Aβ1-40 (Product number, A2326; Sigma Chemical, St Louis, MO, USA) was resuspended at a concentration of 1 mmol/L in saline solution. To obtain the aggregated form of Aβ1-40, the peptide solution was placed in an incubator at 37 °C for 48 h. SF, a colorless powder with purity >99%, was obtained from Suzhou Changtong Chemical (Suzhou, China). SB203580 was obtained from Promega Corporation. The enhanced chemiluminescence kit was from Pierce Biotechnology (Rockford, IL, USA). Phospho-p38 MAPK (Thr180/tyr182, #9211), phospho-MKK3/MKK6 (Ser189/207, #9231), phospho-MAPKAPK-2 (Thr334, #3404), phospho-Hsp27(Ser82, #2401), caspase-3 (#9662), caspase-9 (Human Specific, #9502), caspase-7 (#9492), PARP (#9542), cleaved PARP (ASP330, #9541), and anti-rabbit IgG, HRP-linked antibodies and biotinylated protein ladder detection pack (#7727) were purchased from Cell Signaling (Beverly, MA, USA). IL-1β antibody was obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). β-actin antibody was from Sigma Chemical. SeeBlue Plus2 Pre-stained Standard (Catalog N
Animals and drug treatment Sprague-Dawley rats, weighing 200 g to 220 g, were used in these studies (Grade II, Certificate N
The rats were anesthetized with chloral hydrate (300 mg/kg) and placed in a stereotaxic apparatus. Drugs or vehicles were intracerebroventricularly injected into the animals with a Hamilton microsyringe. The injection lasted 5 min and the needle with the syringe was left in place for 2 min after the injection for the completion of the drug infusion.
In the Aβ1-40 group, the rats were injected with 5 µL Aβ1-40. In the Aβ1-40+SF group, the rats were administered with SF ig (100 mg/kg and 200 mg/kg, daily) for 3 weeks prior to Aβ1-40 injection. The rats in the control group for Aβ1-40 and SF were injected with 5 µL saline solution. In the SB203580 group, the rats were injected with 5 µL SB203580 (8 µg/rat). In the Aβ1-40+SB203580 group, the rats were injected with SB203580 and then with Aβ1-40, 1 h after SB203580 injection. The rats in the SB203580 control group were injected with 5 µL 1% DMSO.
The rats were killed by decapitation 6 h after injection with the drugs or vehicles. Hippocampal slices (500-µm thick) were prepared and immediately frozen on dry ice. The CA1 region was microdissected using a method described in a previous study[21] for Western blot. Seven days after injection, Nissl staining was used to observe the morphological change in the hippocampal CA1 regions. Animals (5 in each group) used for Nissl staining were anesthetized and perfused transcardially with 4% paraformaldelyde.
Western blot analysis Western blot was performed for the analysis of IL-1β, MKK3/MKK6, p38 MAPK, MAPKAPK-2, Hsp27, caspase-9, caspase-7, PARP, and caspase-3. The fresh hippocampal CA1 region was homogenized in RIPA buffer [1% Triton, 0.1% SDS, 0.5% deoxycholate, 1 mmol/L EDTA, 20 mmol/L Tris (pH 7.4), 150 mmol/L NaCl, 10 mmol/L NaF, 0.1 mmol/L PMSF]. The nuclear fractions were first isolated by centrifuging the homogenates at 7500×g at 4 °C for 30 min. The supernatant was further centrifuged at 12 000×g at 4 °C for 20 min to remove insoluble materials. Protein concentrations were quantified by the method of Lowry. Tissue samples were equalized for protein concentration. Proteins were resolved by 10%–12% SDS-PAGE, transferred onto nitrocellulose membranes. Gels were also loaded with colored molecular weight markers to assess electrophoretic transfer, and biotinylated protein ladder marker to estimate molecular weights of bands of interest. The membranes were blocked with 3% BSA in TBS (pH 7.6) for 1 h and incubated overnight at 4 °C with suitably diluted primary antibodies. After extensive washing with TTBS, the membranes were incubated with anti-rabbit IgG, HRP-linked antibody plus anti-biotin antibody for 1 h at room temperature. The blots were detected using the enhanced chemiluminescence (ECL) reaction. After visualization by ECL, all of the nitrocellulose strips were reprobed with β-actin antibody to ensure equal loading of protein on all SDS-PAGE gels. Immunoreactive blots were incubated with alkaline phosphatase-conjugated anti-mouse IgG antibody for 1 h. Finally, the blots were developed with the alkaline phosphatase substrate O-dianisidine tetrazotized along with β-naphthyl acid phosphate. Quantification of protein bands was achieved by densitometric analysis using Chem image 5500 software (UVP, USA).
Nissl staining Seven days after injection of Aβ1-40, the rats (five rats of each group) were perfused transcardially with 4% paraformaldehyde in PBS. The brains were post-fixed for 24 h and were embedded in paraffin. Serial coronal sections (5-µm thickness) were taken from various sections of the brain, stained for Nissl body using cresyl violet, and examined for pathological changes.
To assess hippocampal injury, the number of neurons in the pyramidal layer of the medial CA1 region was counted under a light microscope at 400 magnification according to the method described by Zhang et al[22]. Two continuous fields in hippocampal CA1 subregion were selected for each section and the neurons were counted. The mean of two fields was taken as the neuron number of this section and the mean of four sections was taken as the neuron number of this specimen.
Statistical analysis All data were presented as mean±SD. Statistical analysis was carried out with one-way ANOVA, followed by LSD’s post hoc test, which was provided by SPSS 11.5 statistical software. The level of significance was accepted as P<0.05.
Results
Sodium ferulate inhibited the amyloid-β1-40-induced increase in phospho-MKK3/MKK6 and phospho-p38 MAPK expression As shown in Figure 1A, basal levels of hippocampal phospho-MKK3/MKK6 were very low. Intracerebroventricular injection of preaggregated Aβ1-40 led to a significant increase in phospho-MKK3/MKK6 protein expres-sion. The densitometric analysis revealed that phospho-MKK3/MKK6 level was significantly increased (5.07±0.63-fold relative to control value). SF (100 mg/kg and 200 mg/kg) significantly inhibited Aβ1-40-induced increase in phospho-MKK3/MKK6 expression. However, the selective inhibitor of p38 MAPK, SB203580 (8 µg/rat) did not prevent the increase in phosphorylated MKK3/MKK6 induced by Aβ1-40 (Figure 1A). Surprisingly, phosphorylation of the substrate(s) of activated MKK3/MKK6, p38 MAPK, was increased by Aβ1-40 to a smaller extent. The phospho-p38 MAPK level was modestly elevated (1.51±0.155-fold relative to control value). The Aβ1-40-induced increase in activation of p38 MAPK was completely prevented by SF (100 mg/kg and 200 mg/kg). SB203580 did not prevent an Aβ1-40-induced increase in phospho-p38 MAPK expression (Figure 1B).

Effects of sodium ferulate on amyloid-β1-40-induced phosphorylated MAPKAPK -2 and phosphorylated Hsp27 expressions It is well established that MAPKAPK-2 is phosphorylated and activated by p38 MAPK, and therefore, the effect of Aβ1-40 on its phosphorylation state was investigated in the rat hippocampus. As shown in Figure2A, phosphorylated MAPKAPK-2 in hippocampal CA1 prepared from Aβ1-40-treated rats was significantly reduced compared to that in control rats. This Aβ-induced decrease in phosphorylated MAPKAPK-2 was partly reversed by SF (100 mg/kg and 200 mg/kg). SB203580 (8 µg/rat), the p38 MAPK selective inhibitor, completely inhibited the MAPKAPK-2 phos-phorylation.
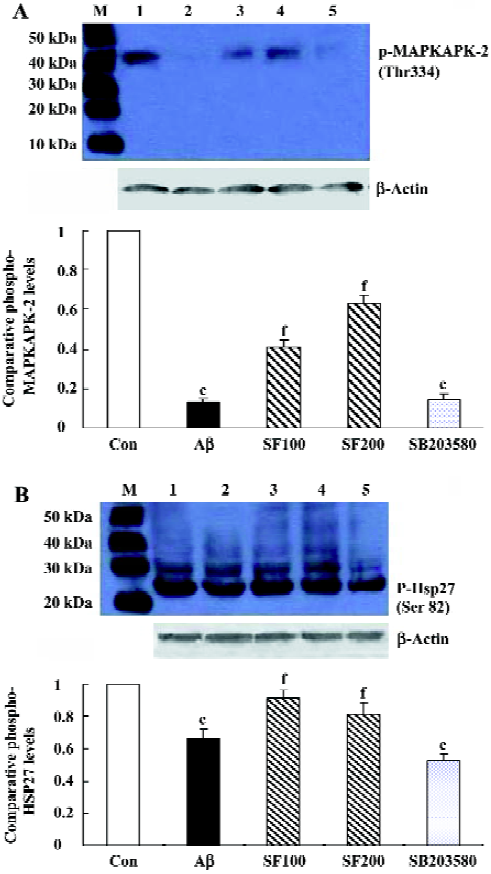
The phosphorylation state of Hsp27 was assessed by immunoblot using a rabbit polyclonal antibody that detects the phosphorylated Hsp27 at Ser82. The results showed that intracerebroventricular injection of Aβ1-40 led to the decrease in Hsp27 phosphorylation, being consistent with the change of MAPKAPK-2 phosphorylation induced by Aβ1-40. SF (100 mg/kg and 200 mg/kg) partly reversed the effect of Aβ1-40 on phosphorylated Hsp27. In addition, SB203580 significantly inhibited Hsp27 phosphorylation (Figure 2B). All the above results support the conclusion that in rat hippocampus the phosphorylation of Hsp27 is catalysed by MAPKAPK-2, which is one of the p38 MAPK substrates. However, in SB203580 control group, 1% DMSO did not show significant effects on phosphorylated MAPKAPK-2 and Hsp 27 expressions compared with the saline solution group (data not shown).
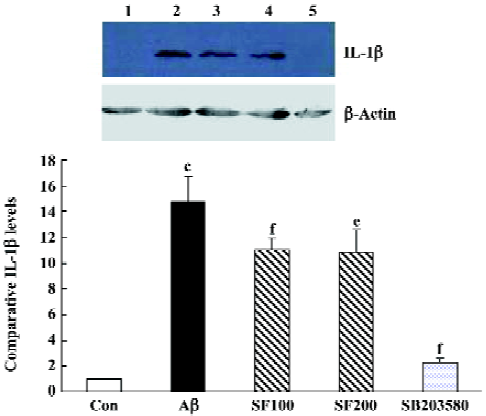
Sodium ferulate and SB203580 inhibit the amyloid-β1-40-induced increase in IL-1β protein level The sample immunoblot and mean data in Figure 4 indicated that the expression of IL-1β in hippocampal samples prepared from Aβ-treated rats was significantly increased compared to samples from control rats. SF (100 mg/kg and 200 mg/kg) markedly inhibited an Aβ-induced increase in IL-1β expression. SB303580 completely abolished an Aβ-induced increase in IL-1β expression, suggesting that Aβ-induced increase in IL-1β expression in the hippocampus is mediated through p38 MAPK pathway (Figure 3).

Sodium ferulate attenuates the amyloid-β1-40-induced caspase cascades and PARP cleavage To test the influence of SF on Aβ1-40-induced neurotoxicity, the expressions of procaspase-9, procaspase-3, procaspase-7, and their cleavage products were analyzed by immunoblotting. The results showed that intracerebroventricular injection of preaggregated Aβ1-40 led to the processing of inactive procaspase-9 into their active forms. Procaspase-9 proteolysis was confirmed by the increase of a 37 kDa fragment in hippocampal CA1. SF (100 mg/kg and 200 mg/kg) significantly prevented Aβ1-40-induced procaspase-9 cleavage. However, SB203580 did not inhibit Aβ1-40-induced pro-caspase-9 cleavage (Figure 4A). According to the picture of the apoptotic pathway, caspase-9 activity is responsible for procaspase-3 and procaspase-7 activation (executioner caspases) by proteolytic cleavage. Procaspase-3 and procaspase-7 processing was investigated by Western blotting. Aβ1-40 induced procaspase-3 processing and caspase-3 activation, as demonstrated in Figure 4B by the appearance of p19 fragments. Similarly, caspase-7 was also cleaved to its p19 active form (Figure 4C) in Aβ-treated rat hippocampus. Both SF (100 mg/kg and 200 mg/kg) and SB203580 inhibited caspase-3 and caspase-7 activation induced by Aβ1-40. Altogether, these observations indicated that SF interfered with the activation of three procaspases: procaspase-9, -3, and -7.
During apoptosis, PARP is one of the earliest targets for caspase-3 cleavage which results in the formation of an 89 kDa C-terminal fragment containing the catalytic domain and a 24 kDa fragment that binds DNA ends[23]. As shown in Figure 4D, its expression level was significantly lower in hippocampal CA1 region prepared from Aβ-treated animals and was associated with the appearance of the 89 kDa fragment of PARP. In SF-treated animals, the expression level of intact PARP (116 kDa) was higher while the expression of the 89 kDa fragment was lower when compared with that of Aβ-treated animals. SB203580 significantly prevented Aβ-induced PARP cleavage (Figure 4D).
Effect of sodium ferulate on the amyloid-β1-40-induced morphological change and number of hippocampal CA1 pyramidal neurons The arrangement of hippocampal CA1 pyramidal neurons of the control group was clearly discernible (Figure 5A). The arrangement of hippocampal pyramidal neurons of Aβ1-40-treated group was sparse and the Nissl body was decreasing or dissolving (Figure 5B). The arrangements of pyramidal neurons of Aβ+SF (100 and 200 mg/kg) groups were better than that of the Aβ1-40-treated group (Figure 5C, 5D). The number of hippocampal CA1 pyramidal neurons of the Aβ1-40-treated group (32±6, n=5) was significantly less than that of the control group (69±3, n=5) and SF 100 mg/kg (66±5, n=5) and 200 mg/kg (72±10, n=5). No significant difference was detected between the control group and SF groups.

Discussion
We report here that Aβ1-40 induced an increase in phosphorylated MKK3/MKK6 and p38 MAPK expressions in hippocampal tissue. This increase, in combination with enhanced IL-1β protein expression, mediated the Aβ-induced activation of the pro-apoptotic pathways, the caspases. SF significantly prevented an Aβ-induced increase in MKK3/MKK6, p38 MAPK and IL-1β. Similarly, SF remarkably inhibited Aβ-induced activation of procaspase 9 and the subsequent procaspase 3 and procaspase 7, and cleavage of PARP. In addition, one of the most interesting aspects of the present results was the discrepancy between the changes in p38 MAPK and the corresponding phospho-MAPKAPK-2. It is obvious from Figure 1B and Figure 2 that intracerebroventricular injection of Aβ1-40 elevated phospho-p38 MAPK expression, but reduced its substrates, phospho-MAPKAPK-2 and phospho-Hsp27 protein expression. SF reversed Aβ1-40 induced these changes. In other words, SF significantly prevented Aβ1-40-induced decrease in phospho-MAPKAPK-2 and phospho-Hsp27. Our evidence indicates that SF may exert its neuroprotective effect by decreasing activation of caspase-9,-3, and -7 and PARP cleavage.
Activation of p38 MAPK is involved in neuronal response to various stresses[24], and this kinase is closely related to hyperphosphorylated tau protein in AD[25]. Our result and previous studies demonstrated that Aβ-induced increase in p38 MAPK activation was accompanied by the increase in IL-1β[26]. SB203580, a selective p38 MAPK inhibitor, completely abolished Aβ-induced increase in IL-1β protein expression, indicating that Aβ-induced increase in IL-1β was mediated through p38 MAPK pathway. Over-expression of IL-1β observed in the AD brain contributes to the neuronal dysfunction and loss characteristic of AD, particularly those involved in formation of neurofibrillary tangle and loss of synapse. In addition, IL-1β upregulates expression and stimulates the processing of the Aβ precursor protein, resulting in amyloidogenic fragments in neurons[27]. In this way, IL-1β may sustain and enhance plaque formation. Alternatively, Aβ, phosphotau or IL-1β may be the stressors of p38 MAPK/JNK. Positive feedback loops may be present in the AD brain whereby the initial stressor is amplified via MAPK pathway activation. SF can inhibit p38 MAPK and IL-1β production, thereby blocking this vicious cycle.
Downstream of p38 MAPKs, there is diversification and extensive branching of signaling pathways. One of the p38 MAPK substrates is MAPKAPK-2, which phosphorylates Hsp27[8]. Our results also demonstrated that SB203580 completely prevented phospho-MAPKAPK-2 protein expression, but partly inhibited phospho-Hsp27 expression, suggesting that in rat hippocampus the upstream of MAPKAPK-2 is p38 MAPK, while the downstream is Hsp27.
In this study, we investigated the effect of Aβ1-40 on phosphorylated MAPKAPK-2 and Hsp27 and effect of SF. Surprisingly, our results showed that intracerebroventricular injection of Aβ1-40 reduced phosphorylated MAPKAPK-2 and Hsp27 protein expressions. SF significantly prevented Aβ1-40-induced decrease in phosphorylated MAPKAPK-2 and Hsp27 protein expression. This might be the other mechanism by which SF-inhibited Aβ1-40 induced the activation of the pro-apoptotic pathways, caspases, of hippocampal neurons. In recent years, evidence has accumulated to show that Hsp27 has cellular protection of the central nervous system. Hsp27 acts via two mechanisms to confer cellular protection. First, as molecular chaperones, Hsp27 are active in the formation and maintenance of the native conformation of cytosolic protein[28] and stabilization of the actin filaments, which make up the cytoskeleton of the cell[29]. Second, Hsp27 functions in neuroprotection through anti-apoptotic actions, particularly on the mitochondrial pathway of caspase-dependent cell death. One of the main mechanisms of caspase activation involves the release of cytochrome c from mitochondria. Cytochrome c interacts and binds with the Apaf-1 resulting in an apoptosome. The apoptosome recruits and activates procaspase-9, which recruits, cleaves and activates caspase-3 and caspase-7. It is these two caspases that mediate the death of the cell through selective proteolysis[30]. Hsp27 inhibits apoptosis by modulating a component of this pathway. Hsp27 inhibits the release of cytochrome c[31] and the formation of a functionally competent apoptosome. Hsp27 binds with cytochrome c after its release from the mitochondria to prevent its interaction with Apaf-1[32]. Hsp27 acts to prevent the activation of caspase-3 directly or via a mechanism similar to Bcl-2, which delays PARP cleavage and procaspase-3 activation[33,34]. Therefore, this has led to the hypothesis that Hsp27 may be useful in the treatment of neurodegenerative diseases.
In summary, our finding indicates that SF reduces caspase-9, -3, and -7 expression and activity and PARP cleavage. This inhibitory effect of SF on the activation of the pro-apoptotic pathways, the caspases, might occur through reduction of inflammatory cytokine IL-1β production and p38 MAPK activity and an increase of Hsp27 expression in rat hippocampus.
References
- Yankner BA. Mechanisms of neuronal degeneration in Alzheimer’s disease. Neuron 1996;16:921-32.
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging 2000;21:383-421.
- Combs CK, Karlo JC, Kao SC, Landreth GE. β-Amyloid stimulation of microglia and monocytes results in TNF-α-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J Neurosci 2001;21:1179-88.
- Minogue AM, Schmid AW, Fogarty MP, Moore AC, Campbell VA, Herron CE, et al. Activation of the c-Jun N-terminal kinase signaling cascade mediates the effect of amyloid-β on long term potentiation and cell death in hippocampus. A role for interleukin-1β? J Biol Chem 2003;278:27971-80.
- Hensley K, Floyd RA, Zheng NY, Nael R, Robinson KA, Nguyen X, et al. p38 kinase is activated in the Alzheimer’s disease brain. J Neurochem 1999;72:2053-8.
- Derijard B, Raingeaud J, Barrett T, Wu IH, Han J, Ulevitch RJ, et al. Independent human MAP-kinase signal transduction pathways defined by MEK and MKK isoforms. Science 1995;267:682-5.
- Stein B, Brady H, Yang MX, Young DB, Barbosa MS. Cloning and characterization of MEK6, a novel member of the mitogen-activated protein kinase kinase cascade. J Biol Chem 1996;271:11427-33.
- Rouse J, Cohen P, Trigon S, Morange M, Alonso-Llamazares A, Zamanillo D, et al. A novel kinase cascade triggered by stress and heat shock that stimulates MAPKAP kinase-2 and phosphorylation of the small heat shock proteins. Cell 1994;78:1027-37.
- Raingeaud J, Gupta S, Rogers JS, Dickens M, Han J, Ulevitch RJ, et al. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J Biol Chem 1995;270:7420-6.
- Concannon CG, Gorman AM, Samall A. On the role of Hsp27 in regulating apoptosis. Apoptosis 2003;8:61-70.
- Scott BC, Butler J, Halliwell B, Aruoma OI. Evaluation of the antioxidant action of ferulic acid and catechins. Free Radic Res Commun 1993;19:241-53.
- Fernandez MA, Saenz MT, Garcia MD. Anti-inflammatory activity in rats and mice of phenolic acids isolated from Scrophularia frutescens. J Pharm Pharmacol 1998;50:1183-6.
- Yan JJ, Cho JY, Kim HS, Kim KL, Jung JS, Huh SO, et al. Protection against beta-amyloid peptide toxicity in vivo with long-term administration of ferulic acid. Br J Pharmacol 2001;133:89-96.
- Kim HS, Cho JY, Kim DH, Yan JJ, Lee HK, Suh HW, et al. Inhibitory effects of long-term administration of ferulic acid on microglial activation induced by intracerebroventricular injection of β-amyloid peptide (1-42) in mice. Biol Pharm Bull 2004;27:120-1.
- Cho JY, Kim HS, Kim DH, Yan JJ, Suh HW, Song DK. Inhibitory effects of long-term administration of ferulic acid on astrocyte activation induced by intracerebroventricular injection of beta-amyloid peptide (1-42) in mice. Prog Neuropsychopharmacol Biol Psychiatry 2005;29:901-7.
- Ono K, Hirohata M, Yamada M. Ferulic acid destabilizes preformed beta-amyloid fibrils in vitro. Biochem Biophys Res Commun 2005;336:444-9.
- Sultana R, Ravagna A, Mohmmad-Abdul H, Calabrese V, Butterfield DA. Ferulic acid ethyl ester protects neurons against amyloid beta-peptide(1-42)-induced oxidative stress and neurotoxicity: relationship to antioxidant activity. J Neurochem 2005;92:749-58.
- Zhang Z, Wei T, Hou J, Li G, Yu S, Xin W. Iron-induced damage and apoptosis in cerebellar granule cells: attenuation by tetra-methylpyrazine and ferulic acid. Eur J Pharmacol 2003;467:41-7.
- Hosoda A, Ozaki Y, Kashiwada A, Mutoh M, Wakabayashi K, Mizuno K, et al. Syntheses of ferulic acid derivatives and their suppressive effects on cyclooxygenase-2 promoter activity. Bioorg Med Chem 2002;10:1189-96.
- Jin Y, Yan EZ, Fan Y, Zong ZH, Qi ZM, Li Z. Sodium ferulate prevents amyloid-beta-induced neurotoxicity through suppression of p38 MAPK and upregulation of ERK-1/2 and Akt/protein kinase B in rat hippocampus. Acta Pharmacol Sin 2005;26:943-51.
- Giovannini MG, Blitzer RD, Wong T, Asoma K, Tsokas P, Morrison JH, et al. Mitogen-activated protein kinase regulates early phosphorylation and delayed expression of Ca2+/calmodulin-dependent protein kinase II in long-term potentiation. J Neurosci 2001;21:7053-62.
- Zhang YM, Yang Q, Xu CT, Li KS, Li WQ. Effects of phenytoin on morphology and structure of hippocampal CA3 pyramidal neurons of rats in chronic stress. Acta Pharmacol Sin 2003;24:403-7.
- Soldani C, Scovassi AI. Poly(ADP-ribose)polymerase-1 cleavage during apoptosis: an update. Apoptosis 2002;7:321-8.
- Kawasaki H, Morooka T, Shimohama S, Kimura J, Hirano T, Gotoh Y, et al. Activation and involvement of p38 mitogen-activated protein kinase in glutamate-induced apoptosis in rat cerebellar granule cells. J Biol Chem 1997;272:18518-21.
- Sheng JG, Jones RA, Zhou XQ, McGinness JM, van Eldik LJ, Mrak RE, et al. Interleukin-1 promotion of MAPK-p38 over-expression in experimental animals and in Alzheimer’s disease: potential significance for tau protein phosphorylation. Neuro-chem Int 2001;39:341-8.
- Giovannini MG, Scali C, Prosperi C, Bellucci A, Vannucchi MG, Rosi S, et al. Beta-amyloid-induced inflammation and cholinergic hypofunction in the rat brain in vivo: involvement of the p38MAPK pathway. Neurobiol Dis 2002;11:257-74.
- Goldgaber D, Harris HW, Hla T, Maciag T, Donnelly RJ, Jacobsen JS, et al. Interleukin 1 regulates synthesis of amyloid beta-protein precursor mRNA in human endothelial cells. Proc Natl Acad Sci USA 1989;86:7606-10.
- Jakob U, Gaestel M, Engel K, Buchner J. Small heat shock proteins are molecular chaperones. J Biol Chem 1993;268:1517-20.
- Guay J, Lambert H, Gingras-Breton G, Lavoie JN, Huot J, Landry J. Regulation of actin filament dynamics by p38 map kinase-mediated phosphorylation of heat shock protein 27. J Cell Sci 1997;110:357-68.
- Franklin TB, Krueger-Naug AM, Clarke DB, Arrigo AP, Currie RW. The role of heat shock proteins Hsp70 and Hsp27 in cellular protection of the central nervous system. Int J Hyperthermia 2005;21:379-92.
- Paul C, Manero F, Gonin S, Kretz-Remy C, Virot S, Arrigo AP. Hsp27 as a negative regulator of cytochrome c release. Mol Cell Biol 2002;22:816-34.
- Bruey JM, Ducasse C, Bonniaud P, Ravagnan L, Susin SA, Diaz-Latoud C, et al. Hsp27 negatively regulates cell death by interacting with cytochrome c. Nat Cell Biol 2000;2:645-52.
- Garrido C, Bruey JM, Fromentin A, Hammann A, Arrigo AP, Solary E. HSP27 inhibits cytochrome c-dependent activation of procaspase-9. FASEB J 1999;13:2061-70.
- Pandey P, Farber R, Nakazawa A, Kumar S, Bharti A, Nalin C, et al. Hsp27 functions as a negative regulator of cytochrome c-dependent activation of procaspase-3. Oncogene 2000;19:1975-81.