
Neuronal nicotinic acetylcholine receptors serve as sensitive targets that mediate β-amyloid neurotoxicity1
Introduction
Alzheimer’s disease (AD) is a syndrome of dementia, characterized by gradual degeneration of basal forebrain cholinergic neurons innervating the cortex, amygdala and hippocampus, which manifests itself through difficulties in maintaining and sustaining attention, and profound cognitive impairments, such as loss of memory and the ability to learn[1−4]. These deficits are thought to be due to selective forebrain cholinergic neuronal degeneration[5]. Thus far, the mechanisms of selective cholinergic neuronal degeneration have been hypothesized to include the impairment of neuronal trophic support, disorders in glucose metabolism or other processes[6], but the precise mechanisms involved are still largely unclear. Recently, accumulating lines of evidence have shown that there is a crucial impairment of nicotinic acetylcholine receptor (nAChR) binding sites in the brain of AD patients[7,8]. β-amyloid peptides (Aβ) directly modulate nAChR function[9−13]. Nicotinic agents have been found to improve cognitive function in AD animal models and AD patients[14−17], suggesting that a relationship exists between nAChRs and Aβ. Therefore, the neuronal nAChR is likely to play an important role in mediating both Aβ toxicity and neural degeneration, and may serve as a therapeutic target for the treatment of AD.
Neuronal nAChRs
Structure and distribution of nAChRs in the central nervous system (CNS)
nAChRs are prototypical members of the ligand-gated ion channel superfamily of neurotransmitter receptors. nAChRs represent both classic and contemporary models for the establishment of concepts pertaining to mechanisms of drug action, synaptic transmission and structural/functional diversity of transmembrane signaling molecules[18−24]. nAChRs are found throughout the nervous system (eg in autonomic and sensory ganglia and the CNS), exist as multiple, diverse subtypes, and are pentamers composed of unique combinations from a family of at least 17 (α1–α10, β1–β4, γ, δ and ε) similar, but genetically-distinct, subunits. Each subunit gene has a unique promoter, even though some genes are clustered, suggesting a means for cell-specific expression. There are also unique protein sequence elements within each subunit, especially in the large, cytoplasmic loop, suggesting a differential post-translational control of subunit trafficking. Most of these nAChR subtypes appear to exist as heteropentamers containing 2 or more different kinds of subunits. For example, heterologous expression studies suggest that α2, α3, α4 or α6 subunits can combine in a binary fashion with β2 or β4 subunits to form ligand-binding and/or functional nAChRs (eg α4β2-nAChRs). β3 and α5 subunits are ‘wild-cards’. They are not able to form functional nAChRs on their own or with any other single type of subunit, but they are capable of integrating into complexes with 2 or more other subunit types to form distinctive trinary or ternary (that also contain more than one of the α2−α4, α6, β2 or β4 subunits found in binary complexes) complexes such as the α4β2α5-nAChR or the α3β2β4α5-nAChR (which is naturally expressed). In contrast, phylogenetically-ancient nAChR α7 subunits are able to form functional homo-pentamers, which constitute the simplest possible prototype for a ligand-gated ion channel. nAChRs containing α7 subunits (α7-nAChR) or α4 and β2 subunits (α4β2*-nAChR) are the most abundant curaremimetic neurotoxin-binding and high affinity nicotine-binding nAChR in the brain. However, other less-abundant nAChRs (eg α3*-nAChR or α6*-nAChR) exist and may also play important roles in brain physiological regulation.
Function of neuronal nAChRs
nAChR function in vertebrate muscle has been comprehensively characterized, and studies of functional nAChRs in autonomic ganglia are rather advanced[23,24]. In regards to nAChRs found centrally, there has been heavy reliance on heterologous expression studies, principally using oocytes as hosts, but the use of transfected mammalian cells has also assisted in defining the realm of possibilities for nAChR subunit compositions that are capable of forming functional, ligand-gated ion channels. Significant insights have been gained about functional nAChRs in the brain from a substantial body of evidence derived using electrophysiological recordings, neurotransmitter release analyses, isotopic ion flux studies and internal calcium ion imaging. Studies using transgenic mice have helped to identify subunits that constitute some native, functional nAChR subtypes[25–30]. Taken collectively, recent findings have indicated that nAChRs in the brain play roles not only in the mediation of classic, excitatory, cholinergic neurotransmission at selected loci, but also and perhaps more globally, in the modulation of neurotransmission of other chemical messengers, including glutamate, γ-aminobutyric acid (GABA), the monoamines dopamine (DA), norepinephrine, serotonin and ACh itself[23,31−33]. This means that some nAChR subtypes have post-synaptic (or peri-synaptic) somatodendritic locali-zations, whereas others have pre-synaptic dispositions. Moreover, some nAChRs have been implicated in processes such as the structuring and maintenance of neurites and synapses[34−36] and even in the modulation of neuronal viability and/or death[37−40]. Therefore, nAChRs play complex and interesting roles in the modulation of chemical milieu in the brain, for the completion of neuronal circuits, and perhaps for the formation and maintenance of synapses. However, more work is required to define functional nAChRs in the CNS and to determine their cellular distributions. Additional studies are also required to determine whether distinctive subunit combinations dictate whether a given nAChR subtype will be positioned pre- or post-synaptically or whether the same nAChR subtype can have either disposition depending on the cellular environment.
As examples, functional nAChRs in the hippocampus, in neurons of the mesocorticolimbic DA system, including the ventral tegmental area (VTA) and substantia nigra pars compacta (SNc), or in forebrain cholinergic neurons, have received attention[41−46]. Some of these functional analyses utilized electrophysiological recordings from brain slices as well as primary cultured hippocampal neurons. Recent exceptions include studies demonstrating the expression in the VTA of functionally distinct nAChRs, including homomeric α7-nAChRs, which are expressed on less than one-half of VTA DAergic neurons, and a variety of non-α7-nAChRs, provisionally identified as α4α6α5(β2)2-, α4α5(β2)2-, α6β2-, and α4β2-nAChRs[26,47,48]. One complication is that the function of some of these putative subtypes has not been convincingly demonstrated, leaving open the possibility that the immunoisolates are not functional nAChRs found on the cell membrane. Interestingly, GABAergic neurons located in the VTA are likely to express relatively simple nAChR subtypes, mainly the α4β2-nAChR, since less than 25% of GABAergic neurons express α3, α5, α6 and β4 subunits[26]. Recently, several research groups have focused their attention on nAChR function in forebrain neurons[49–51] and have found that these nAChRs not only participate in forebrain neuronal function, but are also modulated by Aβ[52]. The diversity in the expression of nAChR subtypes and subunits on different types of neurons located in different brain regions might be the rule more than the exception for other brain regions, but more work is needed before definitive conclusions can be drawn.
Neuronal nAChR changes in AD
The roles of nAChRs in cognitive function and development are well documented[53,54]. Impaired cognition found in AD patients is believed to be correlated with forebrain cholinergic neural degeneration[55,56], and the cholinergic system has been postulated to be the primary target in AD[57,58]. Molecular and neurochemical evidence have indicated that changes in nAChR subtypes occur in the brain of AD patients. Evidence also indicates that a consistent, significant loss of α4-containing nAChRs occurs in a number of neocortical areas and in the hippocampus of patients with AD[59,60]. Cortical α4*-nAChR deficits are significantly correlated with cognitive impairment in AD patients[61,62]. For example, by measuring the binding of specific radiolabeled ligands, such as [3H]epibatidine and [125I]α-bungarotoxin to reflect receptor numbers, a reduction in α4- and α7-nAChR binding sites was found to be associated with AD. On the other hand, the numbers of binding sites reflective of the α4 subtype were significantly elevated in individuals who were habitual smokers[63−65]. However, mRNA levels of different nAChR sub-types, measured either by in situ hybridization or quantitative RT-PCR, were not different when controls and patients with AD were compared[66,67]. At the protein level, α3 and α4 subunit expression in the temporal cortex and hippocampus, and α7 subunit expression in the hippocampus, were significantly lower in AD patients compared to controls[66,68]. Immunohistochemical analyses have shown a significant reduction in the α4 subunit, but not the α7 or α3 subunits, in the brain of AD patients following autopsy compared to age-matched samples[63,69,70]. In summary, the above data suggest that there is a reduction in the number of nAChR binding sites in the brain of patients with AD at the protein level, but not the mRNA level, which implies that the reduction is likely to be due to nAChR post-translational malfunction.
Aβ peptides and neurotoxicity
Aβ and senile plaques
Plaques are defining neuropathological hallmarks of AD and Aβ, the major constituent of plaques, is considered to play an important role in the pathophysiology of AD. Clinical evidence indicates that amyloid plaques are responsible for the pathogenesis of AD[5]. These plaques are mainly composed of the Aβ peptide, which is obtained from an amyloid precursor protein (APP) by proteolytic cleavage and exists in 2 predominant forms: the 40-residue Aβ1–40 and the 42-residue Aβ1–42. Aβ1–40 represents the majority of the Aβ population in normal individuals[25] and Aβ1–42, which exhibits trophic and toxic effects on neurons[5,71], appears to induce the pathogenesis of AD.
Aβ accumulation in AD: in vivo and in vitro studies
A large body of evidence indicates that the accumulation of large intracellular and extracellular aggregates is a histopathological hallmark for the terminal diagnosis of AD. However, it has long been known that the extent of amyloid accumulation does not correlate well with AD pathogenesis[72] and that a significant number of individuals who have not suffered dementia have also shown notable amounts of amyloid plaques. Among in vivo transgenic animals and in vitro cell culture models, pathological changes are frequently observed prior to the onset of amyloid accumulation. These seemingly conflicting lines of evidence can be reconciled by postulating that soluble Aβ, rather than the mature fibrils, represents the primary toxic species in amyloid-associated degenerative disease[73,74]. In AD patients, soluble Aβ correlates better with cognitive decline and loss of synaptic proteins than insoluble, fibril deposits[75,76]. In APP transgenic mouse models, neurological deficits precede the deposition of significant amounts of Aβ, suggesting that the pathophysiology of AD occurs prior to amyloid fibril deposition[77,78].
On the other hand, evidence has also shown heterogeneity in extracellular amyloid in plaques[79]. Contrary to the popularized dogma that all amyloid plaques arise from extracellular deposition, the different forms and magnitudes of amyloid plaques could be the result of multiple mechanisms of formation. For example, it has been proposed that diffuse and dense-core (senile) amyloid plaques differ with respect to glial activity, with the latter primarily being associated with highly reactive microglia[80]. The popular story of extracellular amyloid aggregation which fails to account for the observed heterogeneity in plaques and detected intracellular Aβ together attract more attention to the mechanisms and intracellular aspects of Aβ plaque formation.
Aβ is neurotoxic
The addition of Aβ to cell cultures causes a rapid and large increase in intracellular Ca2+, whereas equivalent amounts of soluble monomer and fibrils have no detectable effects[29,30]. Moreover, Aβ specifically permeabilizes cell membranes. The Ca2+ influx is not blocked by cobalt, indicating that the effect is not due to the activation of existing Ca2+ channels. Aβ also causes leakage of the membrane impermeant dye calcein from cells, indicating that a variety of molecules diffuse across the membrane following Aβ treatment. This conclusion is in agreement with previous studies that reported that Aβ induced the release of dye from phospholipid vesicles[81,82]. It has also been observed in cell cultures that Aβ treatment results in an increase in cytosolic Ca2+ in Ca2+-free medium. This increase can be largely eliminated by pre-treatment with thapsigargin, which depletes endoplasmic reticulum calcium stores[83], suggesting that external application of Aβ leads to the liberation of Ca2+ from intracellular stores. This is consistent with reports that Aβ may penetrate into cells and disrupt intracellular membranes, causing leakage of sequestered Ca2+, but it could also be the consequence of altered intracellular signaling[84]. Under in vivo conditions, the chronic leakage of ions across the plasma membrane may be sufficient to disrupt normal neuronal function and serve as a source of chronic stress that may impair the maintenance of normal membrane potential.
In addition to Ca2+ channel activity, Aβ also seems to activate K+ channels. The mechanism by which Aβ increases K+ current, which results in ensuing neurotoxicity, is unknown, but oxidative stress may be a factor[85,86].
In summary, studies using both in vivo and in vitro preparations indicate that Aβ is neurotoxic and plays a direct role in the pathogenesis of AD.
Aβ modulates nAChRs
Conflicting results
Recent evidence has indicated that nAChRs serve as central targets for Aβ-induced neurotoxic manifestations such as cholinergic hypofunction and cognitive impairment. However, the action of Aβ on nAChRs is not straightforward and there are several discrepancies among different research groups. Some experiments using in vitro preparations suggest that Aβ acts as a nAChR agonist. For example, Aβ1–42 has been shown to activate α7-nAChRs expressed in Xenopus oocytes[11] and native non-α7-nAChRs in acutely dissociated rat basal forebrain neurons[52]. Using isolated pre-synaptic nerve endings from rat hippocampus and neocortex combined with confocal Ca2+ imaging, Aβ1–42 was found to directly evoke a sustained increase in pre-synaptic Ca2+ levels via nAChRs[87]. This action seemed to involve both α7- and non-α7-nAChRs. On the other hand, other groups, including our laboratory, have shown evidence that Aβ acts on nAChRs as an antagonist. Aβ1–42 was shown to block native α7-containing nAChRs in cultured rat hippocampal neurons[10], human α7-nAChRs in Xenopus oocytes[88], rat α4β2-nAChRs in Xenopus oocytes[89], human α4β2-nAChRs in human SH-EP1 cells[90], mouse muscle nAChRs in human kidney BOSC 23 cells[88], Torpedo nAChRs in Xenopus oocytes[89] and non-α7-nAChRs, including α2β2-, α4β2- and α4α5β2-nAChRs in Xenopus oocytes[12]. Recently, a specific model of interaction between Aβ and nAChRs was postulated[31,32]. In addition, a specially designed peptide that binds to Aβ with high affinity has been reported, and interestingly, this peptide virtually abolishes Aβ-induced nAChR inhibition[91]. Therefore, although there are some inconsistencies about the effects of Aβ, which may be explained by the different preparations of Aβ used on different subtypes of nAChRs by different groups, all of the above-mentioned studies prove that Aβ interacts with nAChRs.
Aβ modulates nAChRs: possible mechanisms
There are 2 main features of AD: Aβ protein deposition and severe cholinergic neuronal deficits. Aβ is a 39- to 43-amino acid transmembrane fragment of a large precursor molecule and is found in diffuse and focal deposits throughout the brain in AD patients. It has been shown that the Aβ protein is a major constituent of senile plaque, a neuropathological hallmark of AD and a neurotoxin in various in vivo and in vitro studies. Although the mechanisms by which Aβ causes cholinergic neuronal degeneration are not fully understood, a few hypotheses have been proposed based on current, growing evidence: (1) neuronal death, either by apoptosis or necrosis, primarily occurs in the cholinergic system; (2) insertion of Aβ proteins into the cell membrane destabilizes the membrane and affects its fluidity[92-94]; (3) Aβ affects intracellular Ca2+ homeostasis through either the production of cation ionophores or activation of ligand- and/or voltage-gated channels[95,96], and (4) Aβ affects nAChR function probably through oxidative processes[97,98]. Until now, the precise mechanisms by which Aβ selectively induces degeneration of forebrain cholinergic neurons in AD patients have been unclear.
Aβ modulates nAChRs: homomeric α7-nAChRs
Among nAChRs, the α7 subtype may play the most important role in mediating the toxicity of Aβ. Aβ1–42 binds to α7-nAChRs with a higher affinity compared to Aβ1–40[99]. Therefore, it has been suggested that chronic stimulation of α7-nAChRs by Aβ, mainly by Aβ1–42[11], elevates, at least in part, intracellular Ca2+ levels. It is also involved in the chronic activation of the extracellular signal-regulated kinase (ERK2) isoform of the ERK mitogen-activated protein kinase (MAPK) cascade which leads to the downregulation of MAPK[100]. The ERK2-MAPK signaling pathway plays a critical role in memory formation[29], and its derangement could in part explain the memory impairment observed in patients with AD. Moreover, it has been proposed that downregulation of ERK2-MAPK may be the initial step of a positive-feedback loop that results in Aβ accumulation[100,101]. There is also another explanation implicating the α7-nAChR[102]. Using in vitro preparations, the binding interaction between Aβ1–42, but not Aβ1–40, and α7-nAChRs facilitates internalization and intracellular accumulation of Aβ1–42[102]. Immunohistochemistry and digital imaging studies have revealed that neurons in the brain of AD patients which contain substantial intracellular accumulation of Aβ1–42 invariably express relatively high levels of α7-nAChRs[102,103]. Furthermore, these studies prove the high co-localization of α7-nAChRs and Aβ1–42 within neurons of AD brains. Michael et al introduced a new hypothesis referring to the co-localization of α7-nAChRs and Aβ1–42. They suggested that amyloid plaques may derive from the lysis of forebrain neurons that are overburdened with intracellular accumulation of the α7-nAChR/Aβ1–42 complex, which challenged the prevailing amyloid accumulation story[102,103]. This provides a reasonable explanation for Aβ1–42 causing a reduction in the cell surface-associated α7-nAChR by a relocation of this receptor to intracellular Aβ1–42-positive deposits. This reduction results in the intracellular derangement of calcium cascade, which in turn leads to selective degeneration of cholinergic and cholinoceptive neurons in AD brains.
Another consequence of the interaction between Aβ and α7-nAChRs would be a derangement of the GABA system, which plays a role in long-term potentiation and learning[104]. α7-nAChRs, located on GABAergic interneurons, modulate GABA release, and chronic stimulation of α7-nAChRs would modify GABAergic signaling. Taking these results into consideration, compounds that are able to block the effects of Aβ on α7-nAChR function may possibly be used as therapeutic agents for AD. Moreover, the mechanisms of Aβ-induced damage implicating nAChRs have also been proposed to be involved in the glutamatergic system[104]. By inhibiting glutamate re-uptake by astrocytes, Aβ would promote excessive glutamate stimulation. Glutamate induces an increase in intracellular Ca2+ levels via activation of N-methyl-D-aspartate (NMDA) receptors. This influx of Ca2+ activates nitric oxide (NO) synthase and leads to the production of toxic oxygen radicals and cell death[34]. It has also been reported that α7-nAChR stimulation would promote anti-apoptotic protein synthesis via elevation of intracellular Ca2+ levels and activation of phosphatidylinositol 3-kinase and Akt kinase[105]. These results suggest that α7-nAChR stimulation could be used as a neuroprotective therapy, which could provide the most benefit to patients with AD if the disease is diagnosed in the early stages of development. The above-mentioned different mechanisms suggest that the consequences of α7-nAChR activation, desensitization or inactivation by Aβ1–42 or nAChR agonists, such as nicotine, on AD development are complex or even exhibit opposite effects, suggesting that Aβ modulation of nAChR function is indeed complicated.
Aβ modulates nAChRs: non-α7-nAChRs
A significant decrease in the number of radioligand binding sites corresponding to nAChRs, especially α4-containing nAChRs, is one of the earliest events in the pathogenesis of AD[106], even preceding cholinergic neuronal degeneration. Further support for the cholinergic hypothesis of AD comes from observations that nicotine improves cognitive function in AD patients[107]. Accumulating data also indicates that Aβ1–42 can block non-α7-nAChRs in various neurons or cell lines[92,93].
It has been reported that Aβ can directly modulate α4β2-nAChR function[92,93], which is the most abundant non-7-nAChR subtype in the brain of vertebrates[60,108,109]. Our data have shown that Aβ1–42, at a pathology-relevant concentration (1 nmol/L), can inhibit the human α4β2-nAChR (hα4β2-nAChR) heterologously expressed in human SH-EP1 cells. Aβ1–42-mediated inhibition of hα4β2-nAChR function is non-competitive, voltage-independent and use-independent. This downregulation of hα4β2-nAChR function by Aβ1–42 has been confirmed to not be mediated by nAChR internalization[90]. In addition, we have demonstrated that there is no competition between Aβ1–42, at picomolar to micromolar concentrations, and nAChR agonists based on radioligand binding sites using heterologously expressed hα4β2- or hα7-nAChRs. Therefore, our findings indicate that Aβ1–42 likely acts as a non-competitive antagonist of hα4β2-nAChRs[90].
In Xenopus oocytes expressing various non-α7-nAChRs, including α4β2-nAChRs, Aβ1–42 can reversibly block membrane currents induced by carbachol. More interestingly, altering the α4:β2 RNA ratio of α4β2-nAChRs alters the sensitivity of nAChRs to Aβ1–42. In other words, increasing the relative amount of the α4 subunit significantly decreases the sensitivity of α4β2 channels to Aβ1–42, which suggests that the relative block by Aβ1–42 is affected by the stoichiometry of α4β2 channels[110]. Numerous studies have revealed that Aβ1–42 regulates the function of non-α7-nAChRs. However, links between losses in nAChRs, cholinergic neuronal degeneration and the effects of Aβ have been elusive.
Histological studies showing co-expression of nAChR α7 and β2 subunits in most forebrain cholinergic neurons[111], and heterologous expression work indicating that nAChR α7 and β2 subunits can come together to form heteromeric functional channels[112], suggest that although most α7-nAChRs are formed as homomeric pentamers, others may exist as heteromers, including a possible α7β2-nAChR subtype. However, the expression of functional α7β2-nAChRs in forebrain cholinergic neurons has not been demonstrated and their sensitivity to Aβ has not been determined.
The predominant clinical syndrome associated with AD is a deficiency in both learning and memory capabilities. These deficits are thought to be due to selective forebrain cholinergic neuronal degeneration. Although this selective cholinergic neurodegeneration is largely unclear, several hypotheses have been postulated, including Aβ-induced toxicity, impairment of neuronal trophic support, disorders in glucose metabolism or other processes[113]. The accumulation and aggregation of the Aβ protein in diffuse neuritic plaques is a key pathological hallmark of AD. Aβ accumulation is thought to contribute to cholinergic neuronal degeneration, in turn causing learning and memory deficits[114]. Evidence indicates that Aβ harms central neurons by affecting cellular Ca2+ homeostasis, neurotransmission, neuronal signaling and receptor/ion channel functions[115]. However, most of the relevant experiments have been done using Aβ at concentrations ranging between 100 nmol/L and 10 μmol/L, which are much higher than Aβ concentrations (<5 nmol/L) found in the brain of patients with AD[116,117]. Moreover, the effects of Aβ have been examined in a variety of cell types that may not be appropriate models to characterize the selective effects of Aβ on native forebrain cholinergic neurons.
Figure 1 summarizes the roles of neuronal nAChRs in mediating Aβ-induced neuronal degeneration.
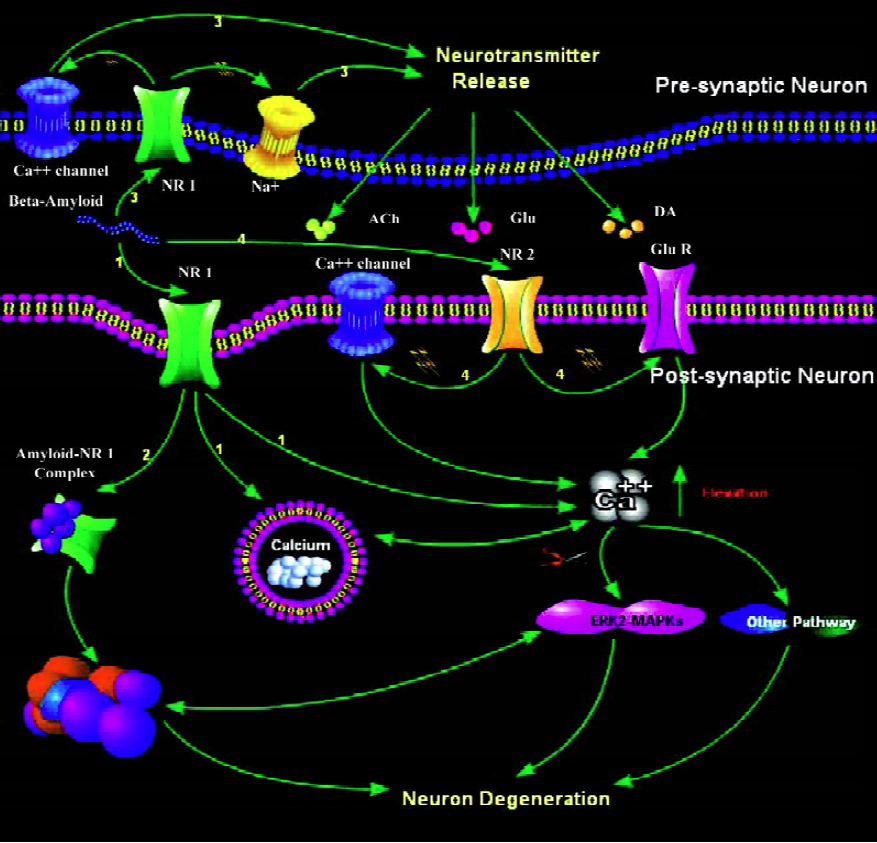
Conclusion
A marked reduction in the number of nAChRs is one of the major neurochemical features of AD in disease-relevant brain regions such as the cortex and hippocampus. This loss is accompanied by a deficiency in the number of forebrain cholinergic neurons, which contributes to the development of cognitive dysfunction. The precise mechanisms that underlie these losses are not yet fully defined. Further development of transgenic models recapitulating these important neurochemical characteristics may help to resolve these issues. Additional major challenges include understanding why aberrant Aβ accumulation occurs, determining if accumulating Aβ is indeed toxic and identifying the precise molecular mechanisms leading to synaptic dysfunction and neuronal degeneration. Such knowledge will help to identify and/or develop novel compounds that can restore cholinergic system function in patients with AD.
Acknowledgement
The authors thank Kevin ELLSWORTH for his assistance in preparing the manuscript.
References
- Coyle JT, Price DL, DeLong MR. Alzheimer’s disease: a disorder of cortical cholinergic innervation. Science 1983;219:1184-90.
- Aubert I, Araujo DM, Cecyre D, Robitaille Y, Gauthier S, Quirion R. Comparative alterations of nicotinic and muscarinic binding sites in Alzheimer’s and Parkinson’s diseases. J Neurochem 1992;58:529-41.
- Selkoe DJ. Alzheimer’s disease results from the cerebral accumulation and cytotoxicity of amyloid β-protein. J Alzheimers Dis 2001;3:75-80.
- Marin DB, Sewell MC, Schlechter A. Alzheimer’s disease. Accurate and early diagnosis in the primary care setting. Geriatrics 2002;57:36-40.
- Auld DS, Kornecook TJ, Bastianetto S, Quirion R. Alzheimer’s disease and the basal forebrain cholinergic system: relations to β-amyloid peptides, cognition, and treatment strategies. Prog Neurobiol 2002;68:209-45.
- Dolezal V, Kasparova J. β-amyloid and cholinergic neurons. Neurochem Res 2003;28:499-506.
- Levin ED, Simon BB. Nicotinic acetylcholine involvement in cognitive function in animals. Psychopharmacology (Berl) 1998;138:217-30.
- Newhouse PA, Kelton M. Clinical aspects of nicotinic agents: therapeutic applications in central nervous system disorders. In Clementi F, Gotti C, Fornasari D, editors. Handbook of experimental pharmacology: neuronal nicotinic receptors. Heidelberg: Springer; 1999. p 779−812.
- Pettit DL, Shao Z, Yakel JL. β-Amyloid (1-42) peptide directly modulates nicotinic receptors in the rat hippocampal slice. J Neurosci 2001;21:RC120.
- Liu Q, Kawai H, Berg DK. β-Amyloid peptide blocks the response of α7-containing nicotinic receptors on hippocampal neurons. Proc Natl Acad Sci USA 2001;98:4734-9.
- Dineley KT, Bell KA, Bui D, Sweatt JD. β-Amyloid peptide activates α7 nicotinic acetylcholine receptors expressed in Xenopus oocytes. J Biol Chem 2002;277:25056-61.
- Lamb PW, Melton MA, Yakel JL. Inhibition of neuronal nicotinic acetylcholine receptor channels expressed in Xenopus oocytes by β-amyloid1-42 peptide. J Mol Neurosci 2005;27:13-21.
- Pym L, Kemp M, Raymond-Delpech V, Buckingham S, Boyd CA, Sattelle D. Subtype-specific actions of β-amyloid peptides on recombinant human neuronal nicotinic acetylcholine receptors (α7, α4α2, α3α4) expressed in Xenopus laevis oocytes. Br J Pharmacol 2005;146:964-71.
- Hernandez CM, Terry AV Jr. Repeated nicotine exposure in rats: effects on memory function, cholinergic markers and nerve growth factor. Neuroscience 2005;130:997-1012.
- Nordberg A, Hellstrom-Lindahl E, Lee M, Johnson M, Mousavi M, Hall R, et al. Chronic nicotine treatment reduces β-amyloidosis in the brain of a mouse model of Alzheimer’s disease (APPsw). J Neurochem 2002;81:655-8.
- White HK, Levin ED. Four-week nicotine skin patch treatment effects on cognitive performance in Alzheimer’s disease. Psychopharmacology (Berl) 1999;143:158-65.
- Seo J, Kim S, Kim H, Park CH, Jeong S, Lee J, et al. Effects of nicotine on APP secretion and Aβ- or CT(105)-induced toxicity. Biol Psychiatry 2001;49:240-7.
- Lindstrom J, Anand R, Gerzanich V, Peng X, Wang F, Wells G. Structure and function of neuronal nicotinic acetylcholine receptors. Prog Brain Res 1996;109:125-37.
- Albuquerque EX, Alkondon M, Pereira EF, Castro NG, Schrattenholz A, Barbosa CT, et al. Properties of neuronal nicotinic acetylcholine receptors: pharmacological characterization and modulation of synaptic function. J Pharmacol Exp Ther 1997;280:1117-36.
- Lukas RJ, Changeux JP, Le Novère N, Albuquerque EX, Balfour DJK, Berg DK, et al. International Union of Pharmacology. current status of the nomenclature for nicotinic acetylcholine receptors and their subunits. Pharmacol Rev 1999;51:397-401.
- Dajas-Bailador F, Wonnacott S. Nicotinic acetylcholine receptors and the regulation of neuronal signalling. Trends Pharmacol Sci 2004;25:317-24.
- Clementi F, Fornasari D, Gotti C. Neuronal nicotinic receptors, important new players in brain function. Eur J Pharmacol 2000;393:3-10.
- Jensen AA, Frolund B, Liljefors T, Krogsgaard-Larsen P. Neuronal nicotinic acetylcholine receptors: structural revelations, target identifications, and therapeutic inspirations. J Med Chem 2005;48:4705-45.
- Lukas RJ. Pharmacological effects of nicotine and nicotinic receptor subtype pharmacological profiles. In: George TP, editor. Medication treatments for nicotine dependence. Boca Raton:CRC Press; 2006. p 7–44.
- Picciotto MR, Zoli M, Rimondini R, Lena C, Marubio LM, Pich EM, et al. Acetylcholine receptors containing the α2 subunit are involved in the reinforcing properties of nicotine. Nature 1998;391:173-7.
- Klink R. de Kerchove, d’Exaerde A, Zoli M, Changeux JP. Molecular and physiological diversity of nicotinic acetylcholine receptors in the midbrain dopaminergic nuclei. J Neurosci 2001;21:1452-63.
- Champtiaux N, Changeux JP. Knockout and knockin mice to investigate the role of nicotinic receptors in the central nervous system. Prog Brain Res 2004;145:235-51.
- Marubio LM, Gardier AM, Durier S, David D, Klink R, Arroyo-Jimenez MM, et al. Effects of nicotine in the dopaminergic system of mice lacking the α4 subunit of neuronal nicotinic acetylcholine receptors. Eur J Neurosci 2003;17:1329-37.
- Tapper AR, McKinney SL, Nashmi R, Schwarz J, Deshpande P, Labarca C, et al. Nicotine activation of α4* receptors: sufficient for reward, tolerance, and sensitization. Science 2004;306:1029-32.
- Salminen O, Murphy KL, McIntosh JM, Drago J, Marks MJ, Collins AC, et al. Subunit composition and pharmacology of two classes of striatal presynaptic nicotinic acetylcholine receptors mediating dopamine release in mice. Mol Pharmacol 2004;65:1526-35.
- McGehee DS, Role LW. Physiological diversity of nicotinic acetylcholine receptors expressed by vertebrate neurons. Annu Rev Physiol 1995;57:521-46.
- Alkondon M, Pereira EF, Barbosa CT, Albuquerque EX. Neuronal nicotinic acetylcholine receptor activation modulates gamma-aminobutyric acid release from CA1 neurons of rat hippocampal slices. J Pharmacol Exp Ther 1997;283:1396-411.
- McGehee DS. Molecular diversity of neuronal nicotinic acetylcholine receptors. Ann N Y Acad Sci 1999;868:565-77.
- Freeman JA. Possible regulatory function of acetylcholine receptor in maintenance of retinotectal synapses. Nature 1977;269:218-22.
- Chan J, Quik M. A role for the nicotinic α-bungarotoxin receptor in neurite outgrowth in PC12 cells. Neuroscience 1993;56:441-51.
- Pugh PC, Berg DK. Neuronal acetylcholine receptors that bind α-bungarotoxin mediate neurite retraction in a calcium-dependent manner. J Neurosci 1994;14:889-96.
- Renshaw GM. [125I]-α-bungarotoxin binding co-varies with motoneurone density during apoptosis. Neuroreport 1994;5:1949-52.
- Hory-Lee F, Frank E. The nicotinic blocking agents d-tubocurare and α-bungarotoxin save motoneurons from naturally occurring death in the absence of neuromuscular blockade. J Neurosci 1995;15:6453-60.
- Renshaw GM, Dyson SE. α-BTX lowers neuronal metabolism during the arrest of motoneurone apoptosis. Neuroreport 1995;6:284-8.
- Treinin M, Chalfie M. A mutated acetylcholine receptor subunit causes neuronal degeneration in C elegans. Neuron 1995;14:871-7.
- Wonnacott S, Drasdo A, Sanderson E, Rowell P. Presynaptic nicotinic receptors and the modulation of transmitter release. Ciba Found Symp 1990;152:87-101.
- Alkondon M, Albuquerque EX. Diversity of nicotinic acetylcholine receptors in rat hippocampal neurons. I. Pharmacological and functional evidence for distinct structural subtypes. J Pharmacol Exp Ther 1993;265:1455-73.
- Alkondon M, Pereira EF, Albuquerque EX. Mapping the location of functional nicotinic and gamma-aminobutyric acid A receptors on hippocampal neurons. J Pharmacol Exp Ther 1996;279:1491-506.
- Dani JA, Radcliffe KA, Pidoplichko VI. Variations in desensitization of nicotinic acetylcholine receptors from hippocampus and midbrain dopamine areas. Eur J Pharmacol 2000;393:31-8.
- Mike A, Castro NG, Albuquerque EX. Choline and acetylcholine have similar kinetic properties of activation and desensitization on the α7 nicotinic receptors in rat hippocampal neurons. Brain Res 2000;882:155-68.
- Dani JA, De Biasi M. Cellular mechanisms of nicotine addiction. Pharmacol Biochem Behav 2001;70:439-46.
- Champtiaux N, Gotti C, Cordero-Erausquin M, David DJ, Przybylski C, Lena C, et al. Subunit composition of functional nicotinic receptors in dopaminergic neurons investigated with knock-out mice. J Neurosci 2003;23:7820-9.
- Wu P, Ma D, Pierzchala M, Wu J, Yang LC, Mai X, et al. The Drosophila acetylcholine receptor subunit D β5 is part of an β-bungarotoxin binding acetylcholine receptor. J Biol Chem 2005;280:20987-94.
- Wu M, Hajszan T, Leranth C, Alreja M. Nicotine recruits a local glutamatergic circuit to excite septohippocampal GABAergic neurons. Eur J Neurosci 2003;18:1155-68.
- Thinschmidt JS, Frazier CJ, King MA, Meyer EM, Papke RL. Septal innervation regulates the function of α7 nicotinic receptors in CA1 hippocampal interneurons. Exp Neurol 2005;195:342-52.
- Thinschmidt JS, Frazier CJ, King MA, Meyer EM, Papke RL. Medial septal/diagonal band cells express multiple functional nicotinic receptor subtypes that are correlated with firing frequency. Neurosci Lett 2005;389:163-8.
- Fu W, Jhamandas JH. α-Amyloid peptide activates non-α7 nicotinic acetylcholine receptors in rat basal forebrain neurons. J Neurophysiol 2003;90:3130-6.
- Granon S, Poucet B, Thinus-Blanc C, Changeux JP, Vidal C. Nicotinic and muscarinic receptors in the rat prefrontal cortex: differential roles in working memory, response selection and effortful processing. Psychopharmacology 1995;119:139-44.
- Levin ED. Nicotinic systems and cognitive function. Psychopharmacology 1992;108:417-31.
- Sugaya K, Giacobini E, Chiappinelli VA. Nicotinic acetylcholine receptor subtypes in human frontal cortex: changes in Alzheimer’s disease. J Neurosci Res 1990;27:349-59.
- Rossner S, Ueberham U, Schliebs R, Perez-Polo JR, Bigl V. The regulation of amyloid precursor protein metabolism by cholinergic mechanisms and neurotrophin receptor signaling. Prog Neurobiol 1998;56:541-69.
- Davies P, Maloney AJF. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 1976;2:1403.
- Perry EK, Gibson PH, Blessed G, Perry RH, Tomlinson BE. Neurotransmitter enzyme abnormalities in senile dementia. Choline acetyltransferase and glutamic acid decarboxylase activities in necropsy brain tissue. J Neurol Sci 1977;34:247-65.
- Court J, Martin-Ruiz C, Piggott M, Spurden D, Griffiths M, Perry E. Nicotinic receptor abnormalities in Alzheimer’s disease. Biol Psychiatry 2001;49:175-84.
- Nordberg A. Nicotinic receptor abnormalities of Alzheimer’s disease: therapeutic implications. Biol Psychiatry 2001;49:200-10.
- Nordberg A, Lundqvist H, Hartvig P, Andersson J, Johansson M, Hellstrom-Lindahi E, et al. Imaging of nicotinic and muscarinic receptors in Alzheimer’s disease: effect of tacrine treatment. Dement Geriatr Cogn Disord 1997;8:78-84.
- Nordberg A, Lundqvist H, Hartvig P, Lilja A, Langstrom B. Kinetic analysis of regional (S)(-)11C-nicotine binding in normal and Alzheimer brains –– in vivo assessment using positron emission tomography. Alzheimer Dis Assoc Disord 1995;9:21-7.
- Martin-Ruiz CM, Court JA, Molnar E, Lee M, Gotti C, Mamalaki A, et al. A4 but not β3 and β7 nicotinic acetylcholine receptor subunits are lost from the temporal cortex in Alzheimer’s disease. J Neurochem 1999;73:1635-40.
- Aubert L, Araujo DM, Cecyre D, Robitaille Y, Gauthier S, Quirion R. Comparative alterations of nicotinic and muscarinic binding sites in Alzheimer’s and Parkinson’s diseases. J Neurochem 1992;58:529-41.
- Hellstrom-Lindahl E, Mousavi M, Zhang X, Ravid R, Nordberg A. Regional distribution of nicotinic receptor subunit mRNAs in human brain: comparison between Alzheimer and normal brain. Brain Res Mol Brain Res 1999;66:94-103.
- Mousavi M, Hellstrom-Lindahl E, Guan ZZ, Shan KR, Ravid R, Nordberg A. Protein and mRNA levels of nicotinic receptors in brain of tobacco using controls and patients with Alzheimer’s disease. Neuroscience 2003;122:515-20.
- Terzano S, Court JA, Fornasari D, Griffiths M, Spurden DP, Lloyd S, et al. Expression of the α3 nicotinic receptor subunit mRNA in aging and Alzheimer’s disease. Brain Res Mol Brain Res 1998;63:72-8.
- Burghaus L, Schutz U, Krempel U, de Vos RA, Jansen Steur EN, Wevers A, et al. Quantitative assessment of nicotinic acetylcholine receptor proteins in the cerebral cortex of Alzheimer patients. Brain Res Mol Brain Res 2000;76:385-8.
- Sparks DL, Beach TG, Lukas RJ. Immunohistochemical localization of nicotinic α2 and β4 receptor subunits in normal human brain and individuals with Lewy body and Alzheimer’s disease: preliminary observations. Neurosci Lett 1998;256:151-4.
- Wevers A, Schroder H. Nicotinic acetylcholine receptors in Alzheimer’s disease. J Alzheimers Dis 1999;1:207-19.
- Zamani MR, Allen YS. Nicotine and its interaction with β-amyloid protein: a short review. Biol Psychiatry 2001;49:221-32.
- Terry RD. The pathogenesis of Alzheimer disease: an alternative to the amyloid hypothesis. J Neuropathol Exp Neurol 1996;55:1023-5.
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, et al. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 1991;30:572-80.
- Klein WL. Aβ toxicity in Alzheimer’s disease: globular oligomers (ADDLs) as new vaccine and drug targets. Neurochem Int 2002;41:345-52.
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, et al. Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann Neurol 1999;46:860-6.
- Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, et al. Soluble amyloid β peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am J Path 1999;155:853-62.
- Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, et al. Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc Natl Acad Sci USA 1999;96:3228-33.
- Westerman MA, Cooper-Blacketer D, Mariash A, Kotilinek L, Kawarabayashi T, Younkin LH, et al. The relationship between Aβ and memory in the Tg2576 mouse model of Alzheimer’s disease. J Neurosci 2002;22:1858-67.
- D’Andrea MR, Cole GM, Ard MD. The microglial phagocytic role with specific plaque types in the Alzheimer disease brain. Neurobiol Aging 2004;25:675-83.
- McGeer PL, Klegeris A, Walker DG, Yasuhara O, McGeer EG. Pathological proteins in senile plaques. Tohoku J Exp Med 1994;174:269-77.
- Anguiano M, Nowak RJ, Lansbury PT Jr. Protofibrillar islet amyloid polypeptide permeabilizes synthetic vesicles by a pore-like mechanism that may be relevant to type II diabetes. Biochemistry 2002; 41: 11 338–43.
- Janson J, Ashley RH, Harrison D, McIntyre S, Butler PC. The mechanism of islet amyloid polypeptide toxicity is membrane disruption by intermediate-sized toxic amyloid particles. Diabetes 1999;48:491-8.
- Mina EW, Demuro A, Kayed R, Milton S, Parker I, Glabe CG. Membrane disruption and elevated intracellular calcium as a common mechanism of amyloid oligomer-induced neurodegeneration. Neuroscience Abstracts 2004;449:20.
- Bucciantini M, Calloni G, Chiti F, Formigli L, Nosi D, Dobson CM, . Prefibrillar amyloid protein aggregates share common features of cytotoxicity. J Biol Chem 2004; 279: 31 374–82.
- Colom LV, Diaz ME, Beers DR, Neely A, Xie WJ, Appel SH, et al. Role of potassium channels in amyloid-induced cell death. J Neurochem 1998;70:1925-34.
- Pike CJ, Balazs R, Cotman CW. Attenuation of β-amyloid neurotoxicity in vitro by potassium-induced depolarization. J Neurochem 1996;67:1774-7.
- Dougherty JJ, Wu J, Nichols RA. β-Amyloid regulation of presynaptic nicotinic receptors in rat hippocampus and neocortex. J Neurosci 2003;30:6740-7.
- Grassi F, Palma E, Tonini R, Amici M, Ballivet M, Eusebi F. Amyloid α1-42 peptide alters the gating of human and mouse β-bungarotoxin-sensitive nicotinic receptors. J Physiol 2003;547:147-57.
- Tozaki H, Matsumoto A, Kanno T, Nagai K, Nagata T, Yamamoto S, et al. The inhibitory and facilitatory actions of amyloid-β peptides on nicotinic ACh receptors and AMPA receptors. Biochem Biophys Res Commun 2002;294:42-5.
- Wu J, Kuo YP, George AA, Xu L, Hu J, Lukas RJ. β-Amyloid directly inhibits human α4 β2-nicotinic acetylcholine receptors heterologously expressed in human SH-EP1 cells. J Biol Chem 2004; 279: 37 842–51.
- Magdesian MH, Nery AA, Martins AH, Juliano MA, Juliano L, Ulrich H, . Peptide blockers of the inhibition of neuronal nicotinic acetylcholine receptors by amyloid β. J Biol Chem 2005; 280: 31 085−90.
- Avdulov NA, Chochina SV, Igbavboa U, O’Hare EO, Schroeder F, Cleary JP, et al. Amyloid β-peptides increase annular and bulk fluidity and induce lipid peroxidation in brain synaptic plasma membranes. J Neurochem 1997;68:2086-91.
- Muller WE, Koch S, Eckert A, Hartmann H, Scheuer K. β-Amyloid peptide decreases membrane fluidity. Brain Res 1995;674:133-6.
- Kanfer JN, Sorrentino G, Sitar DS. Amyloid beta peptide membrane perturbation is the basis for its biological effects. Neurochem Res 1999;24:1621-30.
- Arispe N, Pollard HB, Rojas E. The ability of amyloid β-protein [A β P (1-40)] to form Ca2+ channels provides a mechanism for neuronal death in Alzheimer’s disease. Ann N Y Acad Sci 1994;747:256-66.
- Sanderson KL, Butler L, Ingram VM. Aggregates of a β-amyloid peptide are required to induce calcium currents in neuron-like human teratocarcinoma cells: relation to Alzheimer’s disease. Brain Res 1997;744:7-14.
- Murray IV, Sindoni ME, Axelsen PH. Promotion of oxidative lipid membrane damage by amyloid β proteins. Biochemistry 2005;44:12606-13.
- Tabner BJ, El-Agnaf OM, Turnbull S, German MJ, Paleologou KE, Hayashi Y, . Hydrogen peroxide is generated during the very early stages of aggregation of the amyloid peptides implicated in Alzheimer disease and familial British dementia. J Biol Chem 2005; 280: 35 789–92.
- Wang HY, Lee DH, D’Andrea MR, Peterson PA, Shank RP, Reitz AB. β-amyloid(1-42) binds to α7 nicotinic acetylcholine receptor with high affinity: Implications for Alzheimer’s disease pathology. J Biol Chem 2000;275:5626-32.
- Dineley KT, Westerman M, Bui D, Bell K, Ashe KH, Sweatt JD. β-amyloid activates the mitogen-activated protein kinase cascade via hippocampal α7 nicotinic acetylcholine receptors: In vitro and in vivo mechanisms related to Alzheimer’s disease. J Neurosci 2001;21:4125-33.
- Mills J, Laurent Charest D, Lam F, Beyreuther K, Ida N, Pelech SL, et al. Regulation of amyloid precursor protein catabolism involves the mitogen-activated protein kinase signal transduction pathway. J Neurosci 1997;17:9415-22.
- Nagele RG, D’Andrea MR, Anderson WJ, Wang HY. Intracellular accumulation of β-amyloid(1-42) in neurons is facilitated by the β 7 nicotinic acetylcholine receptor in Alzheimer’s disease. Neuroscience 2002;110:199-211.
- Michael R. D’Andrea, Daniel HSL, Wang HY, Nagele RG. Targeting intracellular Aβ42 for Alzheimer’s disease. drug discovery. Drug Dev Res 2002;56:194-200.
- Alkondon M, Braga MF, Pereira EF, Maelicke A, Albuquerque EX. α7 nicotinic acetylcholine receptors and modulation of GABAergic synaptic transmission in the hippocampus. Eur J Pharmacol 2000;393:59-67.
- Kihara T, Shimohama S, Urushitani M, Sawada H, Kimura J, Kume T, et al. Stimulation of α4β2 nicotinic acetylcholine receptors inhibits β-amyloid toxicity. Brain Res 1998;792:331-4.
- Burghaus L, Schutz U, Krempel U, de Vos RA, Jansen Steur EN, Wevers A, et al. Quantitative assessment of nicotinic acetylcholine receptor proteins in the cerebral cortex of Alzheimer patients. Brain Res Mol Brain Res 2000;76:385-8.
- Levin ED, Rezvani AH. Nicotinic treatment for cognitive dysfunction. Curr Drug Targets CNS Neurol Disord 2002;1:423-31.
- Warpman U, Nordberg A. Epibatidine and ABT 418 reveal selective losses of α4β2 nicotinic receptors in Alzheimer brains. Neuroreport 1995;6:2419-23.
- Rezvani AH, Levin ED. Cognitive effects of nicotine. Biol Psychiatry 2001;49:258-67.
- Wang HY, Lee DH, Davis CB, Shank RP. Amyloid peptide Aβ(1-42) binds selectively and with picomolar affinity to α7 nicotinic acetylcholine receptors. J Neurochem 2000;75:1155-61.
- Azam L, Winzer-Serhan U, Leslie FM. Co-expression of α7 and β2 nicotinic acetylcholine receptor subunit mRNAs within rat brain cholinergic neurons. Neuroscience 2003;119:965-77.
- Khiroug SS, Harkness PC, Lamb PW, Sudweeks SN, Khiroug L, Millar NS, et al. Rat nicotinic ACh receptor α7 and β2 subunits co-assemble to form functional heteromeric nicotinic receptor channels. J Physiol 2002;540:425-34.
- Dolezal V, Kasparova J. β-amyloid and cholinergic neurons. Neurochem Res 2003;28:499-506.
- Selkoe DJ. Translating cell biology into therapeutic advances in Alzheimer’s disease. Nature 1999;399:A23-31.
- Fraser SP, Suh YH, Djamgoz MB. Ionic effects of the Alzheimer’s disease β-amyloid precursor protein and its metabolic fragments. Trends Neurosci 1997;20:67-72.
- Mehta PD, Pirttila T, Mehta SP, Sersen EA, Aisen PS, Wisniewski HM. Plasma and cerebrospinal fluid levels of amyloid β proteins 1-40 and 1-42 in Alzheimer disease. Arch Neurol 2000;57:100-5.
- Kuo YM, Kokjohn TA, Kalback W, Luehrs D, Galasko DR, Chevallier N, et al. Amyloid-β peptides interact with plasma proteins and erythrocytes: implications for their quantitation in plasma. Biochem Biophys Res Commun 2000;268:750-6.