
Suppression of inflammatory cytokine production and oxidative stress by CO-releasing molecules–liberated CO in the small intestine of thermally-injured mice1
Introduction
Extensive burn trauma induces a systemic inflammatory state and oxidative stress that predisposes patients to sepsis and multiple organ failure[1–3]. The activation of a pro-inflammatory cascade plays an important role in the development of major complications associated with burn trauma. Also, it is well known that an acute inflammatory response induced by thermal injury is associated with a variety of systemic alterations, including increased vascular permeability, myocardial dysfunction, hypermetabolism, and altered hepatic synthetic activity[4–6]. The intestine is considered to be the critical organ in the development of organ dysfunction in trauma, burn, and intensive care unit patients[7]. Following thermal injury, the small intestine is subjected to ischemia, and consequently, especially during burn resuscitation, reperfusion injury occurs[8]. Intestinal ischemia–reperfusion results in organ injury through both tissue hypoxia and reperfusion phenomena mediated by inflammatory response and oxidative stress[9,10]. There is increasing evidence that the inflammatory response and oxidative stress in the small intestine exert important roles in the bacteria or endotoxin translocation and the development of SIRS after thermal injury[11–15].
Evidence has indicated that endogenous carbon monoxide (CO), a biproduct of inducible heme oxygenase can modulates inflammation. Many experiments have demonstrated that the administration of exogenous CO inhibits the lipopolysaccharide-induced production of cytokines both in vivo and in vitro, and consequently exhibits important cytoprotective functions and anti-inflammatory properties that are beneficial for the resolution of acute inflammation[16–19].
Recently, transitional metal carbonyls have been identified as potential CO-releasing molecules (CORM) with the potential to facilitate the pharmaceutical use of CO by delivering it to tissues and organs[20]. CORM have been shown to act pharmacologically in rat aortic and cardiac tissues where the liberation of CO produces vasorelaxant effects[21–24] and decreases myocardial ischemia–reperfusion damage[25,26], respectively. Our previous studies[27,28] have shown that the burn-induced overexpression of adhesion molecules [such as Intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1)] on endothelium cells and leukocytes may contribute to liver and lung tissue injury, subsequently leading to the multiple organ dysfunction syndrome. And we also confirmed that CORM-released CO attenuates leukocyte sequestration in the liver, lung, and small intestine of burnt mice by interfering with NF-κB activation and the protein expression of ICAM-1, therefore suppressing the pro-adhesive phenotype of endothelial cells. However, it is still unknown if CORM-released CO can downregulate inflammatory production and oxidative stress in the small intestine after burn injury.
The aim of this paper was to shed light on the role of tricarbonyldichlororuthenium (II) dimer (CORM-2), a novel group of CORM, on the suppression of inflammatory cytokine production and malondialdehyde (MDA) levels as an indicator of the oxidative stress index in the small intestine of thermally-injured mice, and to explore the potential molecular mechanisms.
Materials and methods
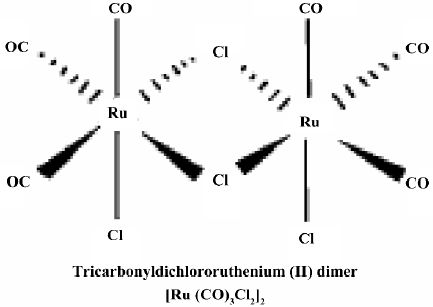
Materials Tricarbonyldichlororuthenium (II) dimer (CORM-2) was obtained from Sigma–Aldrich (St Louis, MO, USA) and solubilized in DMSO to obtain a 10 mmol/L stock. The chemical structure of CORM-2 is represented in Figure 1. The inactive form of the compound (negative control) was also used in some experiments and prepared as follows: CORM-2 was “inactivated” (iCORM)-2 by adding the compound to DMSO and leaving it for 18 h at 37 °C in a 5% CO2 humidified atmosphere to liberate CO. The iCORM-2 solution was finally bubbled with nitrogen to remove the residual CO present in the solution. The inactive form (iCORM-2) was Ru(DMSO)4Cl2, a molecule where the carbonyl groups have been replaced with DMSO. Cell culture reagents were obtained from Gibco (Grand Island, NY, USA), and culture supplies were from Corning (Corning, NY, USA) and Falcon (Lincoln Park, NJ, USA). A polyclonal antibody against inducible nitric oxide (iNOS) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). All other chemicals were of reagent grade and obtained from Sigma–Aldrich (USA) unless otherwise stated.
Animal and burn protocol The C57BL/6 mice (male, n=28, body weight 20±2 g) were fed a standard laboratory diet and water ad libitum. The mice were assigned to 4 groups. The mice in the sham group (n=7) underwent sham thermal injury, whereas the mice in the burn group (n=7) received 15% total body surface area (TBSA) full-thickness thermal injury, the mice in CORM-2 group (n=7) underwent the same thermal injury with immediate administration of CORM-2 (8 mg/kg, iv), and the mice in the burn+iCORM group (n=7) underwent the same thermal injury with immediate administration of iCORM-2 with the same dose as the CORM-2 group. This negative control (iCORM-2) was performed to assess whether the effects observed were due to the CO being liberated by the CORM or caused by other components of the molecules. The concentration of CORM-2 used in the present study was based on a previous report of the use of this compound in mice[29] and preliminary experiments in our laboratory by measuring dynamic COHb levels and peak levels, which averaged 15%±5% above normal levels. The experimental protocol was approved by the Council on Animal Care at Jiangsu University for the protection and the welfare of animals. Under an anesthesia of spontaneous inhalation of isoflurane-N2O (Abbott Laboratories, Missisauga, ON, Canada) in a 60% oxygen–40% nitrogen mixture, the dorsum of each mouse was shaved, and the animal was subjected to 15% TBSA full-thickness thermal injury, as previously described[30,31]. The sham animals were immersed in a room temperature water bath. All animals were resuscitated with 1.5 mL saline immediately after thermal (or sham) injury. No wound care was required for the burn wounds. This burn method achieves a histologically-proven, full-thickness scald injury[32,33]. The animals were killed at 24 h after experimental manipulation.
Collection of sera Blood samples were obtained by cardiac puncture of the left ventricle. The samples were stored in serum tubes (Capiject; Terumo Medical, USA) and immediately centrifuged at 6500×g for 5 min. Serum samples were stored at –80 °C until use.
Preparation of intestinal homogenates Immediately after drawing the blood, the intestine was exposed. Leaving approximately the first 5 cm-long proximal segment of the intestine, 3 cm-long segments of the jejunum and ileum were removed, cleaned, and snap frozen in liquid nitrogen. The samples were stored at –70 °C. Equal weights (100 mg wet weight) of intestine from various groups were suspended in 1 mL phosphate-buffered saline (PBS) and sonicated (30 cycles, twice for 30 s) on ice[34]. Homogenates were cleared by centrifuging at 6500×g at 4 °C, and the supernatants were stored at –70 °C. Protein levels in the homogenates were determined using the Bio-Rad (Hercules, CA, USA) assay kit.
Cell culture and stimulation The human adenocarcinoma cell line Caco-2 was obtained from American Tissue Culture Collection (Rockville, MD, USA). The cells were passaged and grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 1% non-essential amino acids, 10% fetal calf serum, 1000 U/L penicillin, and 1 mg/L streptomycin (complete DMEM). Experiments were carried out with cell monolayers that were 21–25 d old. The cells were seeded in the collagen-coated multiwell dishes (24 pore) with the same cell density. All media were replaced every day when each experiment began. The cells were used between passages 20 and 30. Monolayers were kept at 37 °C, 5% CO2, and 90% related humidity. Caco-2 cell monolayers were stimulated by experimental mouse sera for 4 h and washed, followed by co-incubation with fresh DMEM without serum for another 20 h in 48-well plates. The supernatants of the Caco-2 cells were collected for interleukin (IL)-8 and nitric oxide (NO) level measurements.
Measurement of inflammatory cytokines IL-8, IL-1β, and TNF-α The levels of IL-8 in the supernatants of the Caco-2 cells and the levels of the inflammatory cytokines IL-1β and TNF-α in the ileum and jejunum tissue homogenates were measured using ELISA kits (standard range 1–240, 0.4–300, and 1–1000 pg/mL, respectively; R&D Systems, Minneapolis, MN, USA) following the manufacturer’s instructions.
Measurement of NO in tissue homogenates and supernatants of Caco-2 cells NO was measured by a microplate assay using the Griess reaction with NaNO2 as the standard, which produces a chromophore with the nitrite. Briefly, 100 μL supernatant was removed and incubated with 100 µL Griess reagent (10 μg/L sulfanilamide and 1 μg/L N-1-naphthylethylenediamine dihydrochloride in 25 mL/L phosphoric acid) in a 96-well plate. The plate was incubated for 10 min at room temperature. Nitrite production was quantified spectrophotometrically using an automated colorimetric procedure. Absorbance at 540 nm was measured using a microplate reader (Bio-Tek, USA). The nitrite concentration was calculated by comparing samples with standard solutions of sodium nitrite produced in the culture medium. All samples were assayed in triplicate. Results were expressed as μmol/L.
Measurement of intracellular reactive oxygen species generation in Caco-2 cells Intracellular reactive oxygen species (ROS) generation within Caco-2 cells was assessed by measuring the oxidation of intracellular dihydrorhodamine 123 (DHR 123; Molecular Probes), an oxidant-sensitive fluorochrome, as described previously[35]. Briefly, the cells were treated with DHR 123 (5 mmol/L) for 1 h before being subjected to stimulation of the experimental mouse sera. After 4 h stimulation, the cells were washed with PBS, lysed, and DHR 123 oxidation was assessed spectrofluorometrically at excitation and emission wavelengths of 502 and 523 nm, respectively.
MDA assay MDA formation was utilized as the lipoperoxidation index. The levels of MDA in the tissues were determined following Ohkawa’s method[36]. The ileum and jejunum were homogenized in 1.15% KCl solution (1:10 volume). An aliquot (0.1 mL) of the homogenate was added to a reaction mixture containing 200 μL of 8.1% SDS, 1500 μL of 20% acetic acid (pH 3.5), 1500 μL of 0.8% thiobarbituric acid, and 700 μL distilled water. The samples were then boiled for 1 h at 95 °C and centrifuged at 3000×g for 10 min. The absorbance of the supernatant was measured by spectrophotometry at 532 nm (Jenway, model 6300; Dunmow, Essex, England). Data were expressed in nmol per milligram of wet tissue.
Western blot analysis Tissues were homogenized for extract preparations in ice-cold mild lysis buffer containing 1% Nonidet P-40, 0.15 mol/L NaCl, 0.01 mol/L sodium phosphate (pH 7.2), 2 mmol/L EDTA, 50 mmol/L sodium fluoride, 0.2 mmol/L sodium vanadate, and 1 μg/mL aprotinin. The tissue homogenates were centrifuged at 20 000×g for 15 min and supernatants were collected. SDS–PAGE was performed on equivalent amounts of protein samples using precast 7% resolving/4% stacking Tris–HCl gels (Bio-Rad, USA). Separated proteins were then transferred to polyvinylidene difluoride membranes (Amersham Pharmacia Biotech, Piscataway, NJ, USA). Membranes were blocked in 5% non-fat milk in TBST (TBS buffer containing 0.1% Tween 20) for 1 h at room temperature. Blocked membranes were incubated in primary antibody specific for mouse iNOS at a concentration of 1:2000 dilution in TBST overnight at 4 °C. Then the membranes were washed and probed with a horseradish peroxidase-conjugated secondary antibody (Amersham Pharmacia Biotech, USA) for 1 h at room temperature. Chemiluminescence detection was performed with the Amersham enhanced chemiluminescence detection kit according to the manufacturer’s instructions. To ensure a similar amount of protein in each sample, the membranes were “stripped off,” reprobed with actin, developed with a horseradish peroxidase-conjugated secondary antibody, and visualized by enhanced chemiluminescence.
Statistics Statistical analyses were performed by using SPSS 11.5 software (SPSS, Chicago, IL, USA). Data were expressed as mean±SD. Significant differences between groups with determined with ANOVA. For all analyses, P<0.05 was taken as significant.
Results
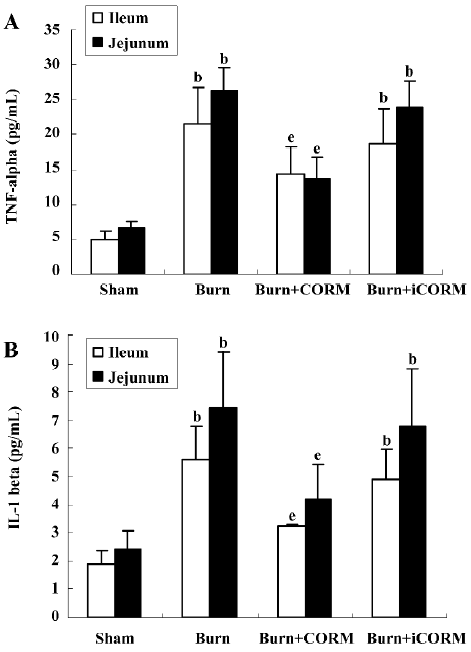
Effect of CORM-2 on TNF-α and IL-1β levels in tissue homogenates of thermally-injured mice The levels of inflammatory cytokines IL-1β and TNF-α in the intestinal tissue were compared among groups. At 24 h after 15% TBSA full-thickness thermal injury, TNF-α and IL-1β levels in tissue homogenates of burn-challenged mice were markedly increased compared to the sham group (P<0.05). After the application of CORM-2, the elevation in tissue homogenate levels of TNF-α and IL-1β were significantly abolished (P<0.05; Figure 2). No significance was shown between the burn group and burn+iCORM group.
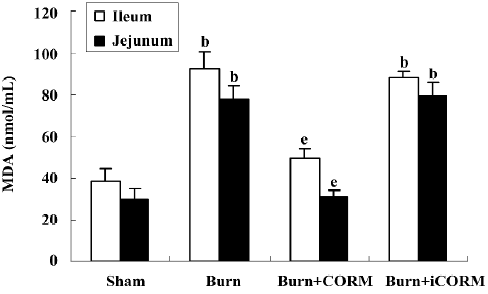
Effect of CORM-2 on tissue MDA in the small intestine of thermally-injured mice Tissue MDA levels are considered important markers of lipoperoxidation associated to oxidative stress. The mean MDA levels detected in the small intestine of mice were significantly affected by thermal injury. At 24 h after 15% TBSA full-thickness thermal injury, the tissue MDA levels in the small intestine significantly increased compared to the sham animals (P<0.05). Following the in vivo administration of CORM-2 (8 mg/kg, iv), tissue MDA levels significantly decreased (P<0.05). No significance was shown between the burn group and burn+iCORM group (Figure 3).
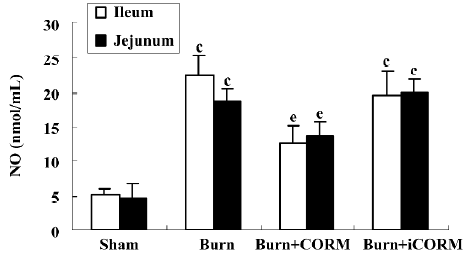
Effect of CORM-2 on NO production in tissue homogenates of thermally-injured mice As shown in Figure 4, the production of NO was low in the sham group. After burn challenge, NO levels in tissue homogenates were significantly increased (P<0.05 compared to the sham group). However, following the administration of CORM-2, NO levels were markedly decreased (P<0.05 compared to the burn group). No significance was shown between the burn group and burn+iCORM group.
Effect of CORM-2 on the expression of iNOS in the small intestine of thermally-injured mice At 24 h after 15% TBSA full-thickness thermal injury, the expression of iNOS in the small intestine significantly increased compared to the sham animals. Following the in vivo administration of CORM-2 (8 mg/kg, iv), the expression of iNOS significantly decreased (Figure 5).
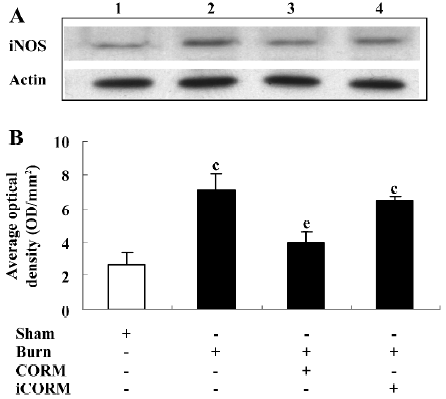
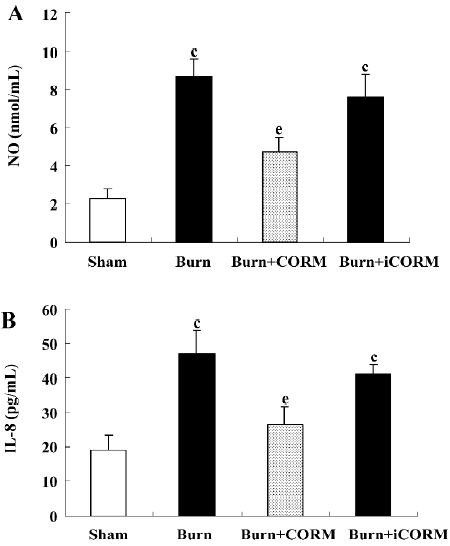
Effect of CORM-2 on NO and IL-8 production in the supernatants of Caco-2 cells stimulated by experimental mouse sera The production of NO and IL-8 were low in the sham mouse serum-stimulated Caco-2 cells. After the stimulation of burnt mouse sera, NO and IL-8 levels were significantly increased (P<0.05 compared to the sham group). The stimulation of Caco-2 cells with CORM-2-treated mouse sera significantly reduced the level of NO and IL-8 generation (P<0.05 compared to the burn group; Figure 6). No significance was shown between the burn group and burn+iCORM group.
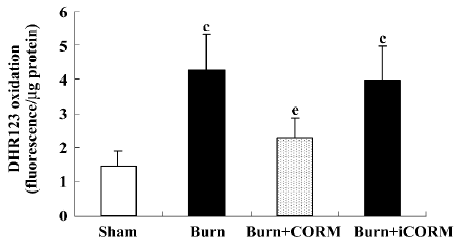
Effect of CORM-2 on the intracellular production of ROS in Caco-2 cells stimulated by experimental mouse sera Intracellular ROS generation within Caco-2 cells was assessed by measuring the oxidation of intracellular DHR 123, an oxidant-sensitive fluorochrome. Burnt mouse sera caused a significant increase of ROS generation in Caco-2 cells; CORM-2 treatment markedly prevented this increase (Figure 7).
Discussion
Severe burns have been known to result in early pathological changes in multiple organs associated with leukocyte sequestration in burnt mice[26,27]. The intestine is one of the most sensitive tissues to ischemia and reperfusion induced by thermal injury. Our previous data have indicated that polymorphonuclear neutrophils (PMN) play an important role in ischemic injury, and reperfusion of the intestine is associated with an accumulation of PMN in the intestinal tissue[37]. We also reported that the CORM-released CO exerts a protective effect against the pathological changes caused by thermal injury of the small intestine. This exogenous CO effectively inhibited the activation of NF-κB and the expression of ICAM-1, suggesting that CORM-2 contributes to the attenuation of leukocyte infiltration to the intestinal tissue after burn challenge[38]. In this study, we employed the same novel metal carbonyl-based compounds (CORM-2) to determine whether they suppress inflammatory cytokine production and oxidative stress in the small intestine of thermally-injured mice.
Pro-inflammatory cytokines/chemokines produced in the intestine play an important role in the pathogenesis of complications of critical illness. One of these, IL-8 has been demonstrated to be a particularly potent chemo-attractant for leukocytes and subsequent inflammation[38]. IL-8 initiates the acute inflammatory cascade and is an early marker of the inflammatory process; its expression must be controlled to prevent excessive tissue injury and damage to local and distal organs[39]. We showed in this study that burnt mouse sera caused a significant increase of IL-8 production in Caco-2 cells, and CORM-2 treatment markedly prevented this increase. These results suggest that the inhibition of IL-8 production by CORM-2 may contribute to the protection of intestinal epithelial cells.
TNF-α is a pleiotropic cytokine with strong pro-inflam-matory, immunomodulatory properties and plays a critical role in inflammation and inflammatory bowel disease[40]. Anti-TNF therapy has been proven to be a milestone in the treatment of inflammatory bowel disease, and equally important in other inflammation-mediated conditions, including viral infections and mucosal inflammation[41–43]. Different strategies have been explored aimed at inhibiting TNF. The present study revealed marked increases in TNF-α levels in the tissue homogenates of the ileum and jejunum after thermal injury. The in vivo administration of CORM-2 was able to inhibit the inflammatory production in enteric tissue induced by thermal injury. Our findings here strongly indicate that CORM-2 appears to inhibit the upregulation of inflammatory production, and consequently might effectively decrease the inflammatory response in the small intestine induced by burns. In parallel, another important cytokine, IL-1β, was also found to be markedly upregulated in the ileum and jejunum of thermally-injured mice. The downregulation of IL-1β levels was most marked in the small intestine of burnt mice treated with CORM-2. These findings demonstrate that treatment with CORM-2 suppresses the production of IL-1β and TNF-α, and subsequently suppresses the inflammatory response induced by burn injury in the small intestine.
Tissue MDA content, the last product of lipid breakdown caused by oxidative stress, is considered to be a good indicator of radical-induced lipid peroxidation[44]. In this study, we found increased intestinal MDA levels in the burnt mice groups, suggesting that in our experimental conditions, after 24 h from manipulation, a significant increase of oxidative stress occurs. The in vivo administration of CORM-2 led to the significant downregulation of mean MDA levels in burnt mice. This indicated that CORM-2 effectively prevents lipid peroxidation in the small intestine after thermal injury, consequently decreases the production of oxidants, and reduces tissue oxidative injury, which contributes to bowel functional damage. Glutathione (GSH), a key antioxidant, is an important constituent of intracellular protective mechanisms against oxidative stress[45]. Among the antioxidant defense mechanisms is GSH, which removes ROS once formed. In the present study, GSH was therefore measured for investigating the antioxidant defense mechanisms in the small intestine of thermally-injured mice. We found that GSH declined significantly in the burn-challenged group (0.10 mg/g) as compared to the control group (0.89 mg/g), while CORM-2 treatment significantly reversed the GSH level reduction (0.48 mg/g, data not shown). Since the administration of CORM-2 prevents intestinal GSH depletion, it appears that the protective effect of CORM-2 involves the maintenance of antioxidant capacity in protecting the intestinal tissue against thermal injury-induced oxidative stress.
NO, an inflammatory factor, is an intercellular signaling factor that is produced by innate immune cells in response to endotoxins and other inflammatory stimuli[46]. Although it is suggested that NO has a counter-inflammatory activity acting on cells of the adaptive immune system, in particular, T cells[47], many studies have well established that NO is indeed a pro-inflammatory mediator. There is also evidence to indicate that NO is an important determinant of the balance between Th1- and Th2-type immune responses[48]. In inflammation and many pathological conditions, the simultaneous cellular production of superoxide anion and NO may occur, potentially leading to the continuous formation of peroxynitrite. Peroxynitrite leads to organ damage, possibly through lipid peroxidation and/or nitration of cell membrane proteins. We previously demonstrated that thermal injury induced lung neutrophil deposition, lung iNOS expression, and lung damage[27]. We have also shown that NO from iNOS regulates pro-inflammatory activation, gene expression, and tissue injury in the liver after thermal injury, and CORM-2 inhibits the expression of iNOS in the liver tissue, reducing liver injury and tissue PMN infiltration in thermally-injured mice[26]. In this study, we measured the NO production and expression levels of iNOS in tissue homogenates of the ileum and jejunum of burnt mice following resuscitation to determine whether iNOS was also generated at this site. Our study here showed that there was a significant increase of intestinal mucosal iNOS activity within the base of intestinal villi following thermal injury, while NO production also markedly increased, suggesting that peroxynitrite plays a vital role in thermal injury-induced intestine damage. The production of NO and the expression of iNOS were significantly inhibited by the in vivo administration of CORM-2. Although future considerations of therapeutic strategies targeting NO or other immune regulatory networks during sepsis and trauma need to consider the balance between Th1- and Th2-type control of inflammatory responses as an important determinant, the present data show that CORM-2 exhibits, at least partly, an important role in the inhibition of iNOS expression, subsequently downregulates NO production, and attenuates oxidative stress and tissue damage. In parallel, the results of the in vitro experiments showed that burnt mouse sera caused a significant increase of NO production and accelerated the generation of intracellular ROS in Caco-2 cells, whereas CORM-2 treatment effectively prevented this increase. This is strong evidence that Caco-2 cells are sensitive to burnt mouse sera, which contain rich inflammatory mediators.
In conclusion, the present study clarifies the role of CORM-2, one of the novel CO-releasing molecules, on the mechanisms of downregulating inflammatory production and suppressing oxidative stress in the small intestine of thermally-injured mice. The application of CORM-2 on burnt mice decreased the production of IL-1β, TNF-α, and IL-8 to control excessive inflammatory responses and prevented the overexpression of iNOS in the small intestine. This was accompanied by a decrease of NO production and ROS generation. Taken together, these findings indicate that CORM-released CO modulates gut inflammation in burnt mice by interfering with the protein expression of iNOS, therefore suppressing oxidative stress and tissue damage. Of course, further studies are now required to understand the detailed mechanisms of the anti-inflammatory effects mediated by CORM and to determine contributions to the development of a therapeutic approach to protect against gut damage during severe burn injury.
Acknowledgement
We are grateful for the technical assistance of Prof Gediminas CEPINSKAS from the Centre for Critical Illness Research, University of Western Ontario, Canada.
References
- Baue AE, Durham R, Faist E. Systemic inflammatory response syndrome (SIRS), multiple organ dysfunction syndrome (MODS), multiple organ failure (MOF): are we winning the battle? Shock 1998;10:79-89.
- Harris BH, Gelfand JA. The immune response to trauma. Semin Pediatr Surg 1995;4:77-82.
- Saffle JR, Sullivan JJ, Tuohig GM, Larson CM. Multiple organ failure in patients with thermal injury. Crit Care Med 1993;21:1673-83.
- Dehne MG, Sablotzki A, Hoffmann A, Muhling J, Dietrich FE, Hempelmann G. Alterations of acute phase reaction and cytokine production in patients following severe burn injury. Burns 2002;28:535-42.
- Cohen J. The immunopathogenesis of sepsis. Nature 2002;420:885-91.
- Brun-Buisson C. The epidemiology of the systemic inflammatory response. Intensive Care Med 2000;26:S64-74.
- Moore EE. Mesenteric lymph: the critical bridge between dysfunctional gut and multiple organ failure. Shock 1998;10:415-6.
- Ward PA, Till GO. Pathophysiologic events related to thermal injury of skin. J Trauma 1990;30:S75-9.
- Li L, Zhang YM, Qiao WL, Wang L, Zhang JF. Effects of hypothalamic paraventricular nuclei on apoptosis and proliferation of gastric mucosal cells induced by ischemia/reperfusion in rats. World J Gastroenterol 2007;13:874-81.
- Liu KX, Wu WK, He W, Liu CL. Ginkgo biloba extract (EGb 761) attenuates lung injury induced by intestinal ischemia/reperfusion in rats: Roles of oxidative stress and nitric oxide. World J Gastroenterol 2007;13:299-305.
- Sener G, Sehirli AO, Satýrog H, Keyer-Uysal M, C, Yeðen B. Melatonin improves oxidative organ damage in a rat model of thermal injury. Burns 2002;28:419-25.
- Horton JW. Free radicals and lipid peroxidation mediated injury in burn trauma: the role of antioxidant therapy. Toxicology 2003;189:75-88.
- Sun Z, Wang X, Lasson AE, Böjesson A, Annborn M, Andersson R. Effects of inhibition of PAF, ICAM-1 and PECAM-1 on gut barrier failure caused by intestinal ischemia and reperfusion. Scand J Gastroenterol 2001;36:55-65.
- Ghandour S, Cetinel S, Kurtel H. Endothelin-3 induced mesenteric vasoconstriction and PMN infiltration in the rat small intestine: role of endothelin receptors. Regulatory Peptides 2004;119:125-31.
- Karabeyoðlu M, Unal B, Bozkurt B, Dolapçi I, Bilgihan A, Karabeyoðlu I, et al. The effect of ethyl pyruvate on oxidative stress in intestine and bacterial translocation after thermal injury. J Surg Res 2008;144:59-63.
- Ko JS, Yang HR, Chang JY, Seo JK. Lactobacillus plantarum inhibits epithelial barrier dysfunction and interleukin-8 secretion induced by tumor necrosis factor-α. World J Gastroenterol 2007;13:1962-5.
- Hayashi S, Takamiya R, Yamaguchi T, Matsumoto K, Tojo SJ, Tamatani T, et al. Induction of heme oxygenase-1 suppresses venular leukocyte adhesion elicited by oxidative stress. Role of bilirubin generatedby the enzyme. Circ Res 1999;85:663-71.
- Lee T, Chau LY. Heme oxygenase-1 mediates the anti-inflammatory effect of interleukin-10 in mice. Nat Med 2002;8:240-6.
- Otterbein LE, Soares M, Yamashita K, Bach FH. Heme oxygenase-1: unleashing the protective properties of heme. Trends Immunol 2003;24:449-55.
- Motterlini R, Mann BE, Johnson TR, Clark JE, Foresti R, Green CJ. Bioactivity and pharmacological actions of carbon monoxide-releasing molecules. Curr Pharm Des 2003;9:2525-39.
- Motterlini R, Clark JE, Foresti R, Sarathchandra P, Mann BE, Green CJ. Carbon monoxide-releasing molecules: characterization of biochemical and vascular activities. Circ Res 2002;90:E17-24.
- Motterlini R, Sawle P, Hammad J, Bains S, Alberto R, Foresti R, et al. CORM-A1: a new pharmacologically active carbon monoxide-releasing molecule. FASEB J 2005;19:284-6.
- Johnson TR, Mann BE, Clark JE, Foresti R, Green CJ, Motterlini R. Metal carbonyls: a new class of pharmaceuticals? Angew Chem Int Ed Engl 2003;42:3722-9.
- Foresti R, Hammad J, Clark JE, Johnson TR, Mann BE, Friebe A, et al. Vasoactive properties of CORM-3, a novel water-soluble carbon monoxide-releasing molecule. Br J Pharmacol 2004;142:453-60.
- Clark JE, Naughton P, Shurey S, Green CJ, Johnson TR, Mann BE, et al. Cardioprotective actions by a water-soluble carbon monoxide-releasing molecule. Circ Res 2003;93:2-8.
- Guo Y, Stein AB, Wu WJ, Tan W, Zhu X, Li QH, et al. Administration of a coreleasing molecule at the time of reperfusion reduces infarct size in vivo. Am J Physiol Heart Circ Physiol 2004;286:H1649-53.
- Sun BW, Chen ZY, Chen X, Liu C. Attenuation of leukocytes sequestration by carbon monoxide-releasing molecules: liberated carbon monoxide in the liver of thermally injured mice. J Burn Care Res 2007;28:173-81.
- Sun B, Sun H, Liu C, Shen J, Chen Z, Chen X. Role of CO-releasing molecules (CORM-2)-liberated CO in attenuating leukocytes sequestration and inflammatory responses in the lung of thermally injured mice. J Surg Res 2007;139:128-35.
- Stein AB, Guo Y, Tan W, Wu WJ, Zhu X, Li Q, et al. Administration of a CO-releasing molecule induces late preconditioning against myocardial infarction. J Mol Cell Cardiol 2005;38:127-34.
- Faunce DE, Gregory MS, Kovacs EJ. Effects of acute ethanol exposure on cellular immune responses in a murine model of thermal injury. J Leukoc Biol 1997;62:733-40.
- Gamelli RL, He LK, Liu H. Macrophage suppression of granulocyte and macrophage growth following burn wound infection. J Trauma 1994;37:888-92.
- Stengle J, Meyers R, Pyle J, Dries DJ. Neutrophil recruitment after remote scald injury. J Burn Care Rehab 1996;17:14-8.
- Faunce DE, Llanas JN, Patel PJ, Gregory MS, Duffner LA, Kovacs EJ. Neutrophil chemokine production in the skin following scald injury. Burns 1999;25:403-7.
- Rana SN, Li X, Chaudry IH, Bland KI, Choudhry MA. Inhibition of IL-18 reduces myeloperoxidase activity and prevents edema in intestine following alcohol and burn injury. J Leukoc Biol 2005;77:719-28.
- Cepinskas G, Lush CW, Kvietys PR. Anoxia/reoxygenation-induced tolerance with respect to polymorphonuclear leukocyte adhesion to cultured endothelial cells: a nuclear factor-κB phenomenon Circ Res 1999;84:103-12.
- Ohkawa H, Ohishi N, Yagi K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem 1979;95:351-8.
- Sun BW, Jin Q, Sun Y, Sun ZW, Chen X, Chen ZY, et al. CO liberated from CO-releasing molecules attenuates leukocyte infiltration in the small intestine of thermally injured mice. World J Gastroenterol 2007;13:6183-90.
- Baggiolini M. Chemokines in pathology and medicine. J Intern Med 2001;250:91-104.
- De Dooy JJ, Mahieu LM, Van Bever HP. The role of inflammation in the development of chronic lung disease in neonates. Eur J Pediatr 2001;160:457-63.
- Lin F, Spencer D, Hatala DA, Levine AD, Medof ME. Decay-accelerating factor deficiency increases susceptibility to dextran sulfate sodium-induced colitis: role for complement in inflammatory bowel disease. J Immunol 2004;172:3836-41.
- Van Assche G, Rutgeerts P. Anti-TNF agents in Crohn’s disease. Expert Opin Investig Drugs 2000;9:103-11.
- Kollias G, Kontoyiannis D. Role of TNF/TNFR in autoimmunity: specific TNF receptor blockade may be advantageous to anti-TNF treatments. Cytokine Growth Factor Rev 2002;13:315-21.
- Hirata I, Yasumoto S, Toshina K, Inoue T, Nishikawa T, Murano N, et al. Evaluation of the effect of pyrrolidine dithiocarbamate in suppressing inflammation in mice with dextran sodium sulfate-induced colitis. World J Gastroenterol 2007;13:1666-71.
- Kabaroglu C, Akisu M, Habif S, Mutaf I, Turgan N, Parildar Z, et al. Effects of L-arginine and L-carnitine in hypoxia/reoxygenation-induced intestinal injury. Pediatr Int 2005;47:10-14.
- Ross D. Glutathione, free radicals and chemotherapeutic agents. Mechanisms of free-radical induced toxicity and glutathione-dependent protection. Pharmacol Therap 1988;37:231-49.
- MacMicking J, Xie QW, Nathan C. Nitric oxide and macrophage function. Annu Rev Immunol 1997;15:323-50.
- Van der Veen RC. Nitric oxide and T helper cell immunity. Int Immunopharmacol 2001;1:1491-500.
- Miki S, Takeyama N, Tanaka T. Immune dysfunction in endotoxicosis: Role of nitric oxide produced by inducible nitric oxide synthase. Crit Care Med 2005;33:716-20.