
Niflumic acid hyperpolarizes smooth muscle cells via calcium-activated potassium channel in spiral modiolar artery of guinea pigs1
Introduction
Changing blood circulation in the inner ear is implicated in many physiological and pathological conditions of hearing function. For example, the stimulation of loud sound can cause a significant reduction in cochlear blood flow[1], while anoxia or interruption of cochlear blood flow causes a drastic reduction of cochlear function[2]. Some forms of sudden deafness may also be due to inner ear circulation problems[3,4]. In addition, disturbances of inner ear circulation are associated with increased sensitivity to ototoxic drugs and noise-induced trauma[5,6]. Thus it is very important to study the characteristics of inner ear microcirculation to reveal the physiological function of the smooth muscle cells (SMC) in the cochlear spiral modiolar artery (SMA), which is only an arteriole into the inner ear.
Large-conductance calcium-activated potassium (KCa) channels are present in a variety of cell types[7-12]. In neurons, they may regulate cell firing[13,14], and in smooth muscle, they seem to play an important role in maintaining visceral and vascular tone[15-18]. Many chloride channel inhibitors, including members of the non-steroidal anti-inflammatory drug family, such as niflumic acid (NFA), not only inhibit Cl– conductance[19-21], but also stimulate large-conductance KCa channels in vascular smooth muscle of the rabbit portal vein[22] and pig coronary[23]. In our preliminary study, we found that KCa channels might exist in the SMC and endothelial cells (EC) of the cochlear SMA[12]. Moreover, chloride channel blockers could inhibit excitatory junction potentials in the SMC of the cochlear SMA in guinea pigs[24].
The aim of the present work was to use direct intracellular recordings of membrane potential and conventional whole-cell recordings to investigate the effect of NFA (a chloride channel blocker) on the SMC in the cochlear SMA of guinea pigs. The results suggest that NFA hyperpolarizes SMC by activating KCa channels in the SMA of guinea pigs.
Materials and methods
Animals and SMA preparation Guinea pigs (300–500 g) were anesthetized and then killed by exsanguination[25]. The anesthesia was accomplished by an intramuscular injection of an anesthetic mixture (1 mL/kg) of 500 mg ketamine, 20 mg xylazine, and 10 mg acepromazine in 8.5 mL H2O. Both bullae were rapidly removed and transferred to a Petri dish filled with a physiological solution (Krebs) composed of (in mmol/L): 125 NaCl, 5 KCl, 1.6 CaCl2, 1.2 MgCl2, 1.2 NaH2PO4, 18 NaHCO3, and 8.2 glucose, and saturated with 95% O2 and 5% CO2 at 35 ℃ (pH 7.4). The SMA and some related radiating arterioles were further dissected from the cochlea under a dissecting microscope. The vascular preparation was incubated for 0.5–24 h in the physiological solution and then transferred to a recording bath. A 2-5 mm long segment of the SMA was cleaned free of spongy connective tissues and pinned with minimum nails to the silicon rubber layer (Sylgard 184, Dow Corning, USA) in the bottom of an organ bath (volume 0.5 mL) and continuously perfused with a 35 ℃ Krebs solution. When needed, a high potassium Krebs solution was made by additional KCl and accordingly-reduced NaCl.
Intracellular recording Intracellular recording was made from a segment of the SMA in the basal and the second turn of the cochlea, as described previously[25,26]. Briefly, the micro-electrode was filled with 2 mol/L KCl, with a tip resistance of 60–150 MΩ. Intracellular penetration was obtained by advancing the electrode into the adventitial surface of the vessel. The transmembrane potentials and current were simultaneously monitored by an Axoclamp 2B preamplifier (Axon Instruments, Burlingame, CA, USA).
The electrical signals were recorded on a pen recorder and a personal computer equipped with Axoscope8 and pClamp6 software (Axon Instruments, USA) using sampling intervals of 0.1, 0.5, or 10 ms. The resting potential was usually determined 5 min after the initial voltage jump at penetration, and checked by the voltage jump at the withdrawal of the electrode. The input resistance was measured by applying 0.5 nA 0.5–1 s current pulses via the recording electrode with the bridge balance well adjusted on the preamplifier[26].
Tight-seal whole-cell recording Using the Axopatch 200B amplifier (Axon Instruments, Union City, CA, USA), conventional whole-cell recordings were performed on smooth muscle cells in situ from the SMA[27]. The specimen was continuously superfused with the normal external solution (0.2 mL·min–1) at room temperature (22–25 ℃). Recording pipettes were made of borosilicate glass capillaries with filament (OD 1.5 mm, ID 0.84 mm; World Precision Instruments, Sarasota, FL, USA) and pulled by a P-80 puller (Sutter Instruments, USA). The pipette was filled with an internal solution containing (in mmol/L): 130 KCl, 10 NaCl, 2 CaCl2, 1.2 MgCl2, 10 HEPES, 5 EGTA (118 nmol/L free Ca2+), and 7.5 glucose, and adjusted to pH 7.2 and to osmolarity 290 mOsm/L. The recording pipettes had a tip of ~1 μmol/L OD and a resistance of ~5 MΩ. Pipette capacitance was well compensated, while membrane input capacitance uncompensated to monitor access resistance and membrane parameters online. The voltage clamping error introduced by the current passing the access resistance was corrected offline according to the equation Vm=Vc-IRa (where Vm is the actual clamping membrane voltage and Vc is the apparent command voltage), except where noted otherwise. Leak subtraction was done offline when appropriate. The membrane current or voltage signal was low-pass filtered at 5 or 10 kHz (–3 dB); data were recorded on a personal computer equipped with a Digidata 1322A AD-interface and pClamp 9.2 software (Axon Instruments, USA) at a sampling interval of 10, 20, or 100 ms. A gap-free recording was simultaneously carried out by a Minidigi digitizer and Axoscope 9.2 software (Axon Instruments, USA) at a sampling interval of 50 ms.
Drugs application Drugs in known concentrations were applied via the bathing solution. The solution passing the recording chamber could be switched, without change in the flow rate or temperature, to one that contained a drug or one that was of different ionic composition. The drugs used in this study were: NFA, indanyloxyacetic acid 94 (IAA-94), disodium 4,4’-diisothiocyanatostilbene-2,2’-disulfonate (DIDS), charybdotoxin (ChTX), iberiotoxin (IbTX), 1,2-bis(2-aminophenoxy) ethane-N,N,N’,N’-tetraacetic acid tetrakis acetoxymethyl ester (BAPTA-AM), tetraethylammonium (TEA), 4-aminopyridine (4-AP), barium, glipizide, apamin, CdCl2, and ouabain (all from Sigma Research Biochemicals, St Louis, MO, USA).
Statistical analysis The values of NFA-induced responses were expressed as mean±SEM and compared with t-test.
Results
Effects of NFA, IAA-94, and DIDS on SMC in SMA The resting potentials (RP) of SMC in the SMA were measured in a normal Krebs solution (5 mmol/L potassium) with 5–10 min duration after the cell was penetrated and the membrane potential level became stabilized. We previously reported that SMC have 2 stable but mutually-convertible (Figure 1B,2A,4A), levels of RP, that is, 1 was near -45 mV and the other was approximately –75 mV, termed as low and high RP, respectively[26]. So under the same condition, stable intracellular recordings were successfully made in more than 210 cells randomly penetrated along the segments of the proximal half of 77 SMA from either side. The recording lasted from 5 min to 6.5 h. The mean RP were –75.46±0.59 mV (n=87) and –40.66±0.41 mV (n=123) in the high and low RP cells, respectively. The mean RP showed no difference from our preliminary study result (–74±0.46 mV and –41±0.52 mV)[26].
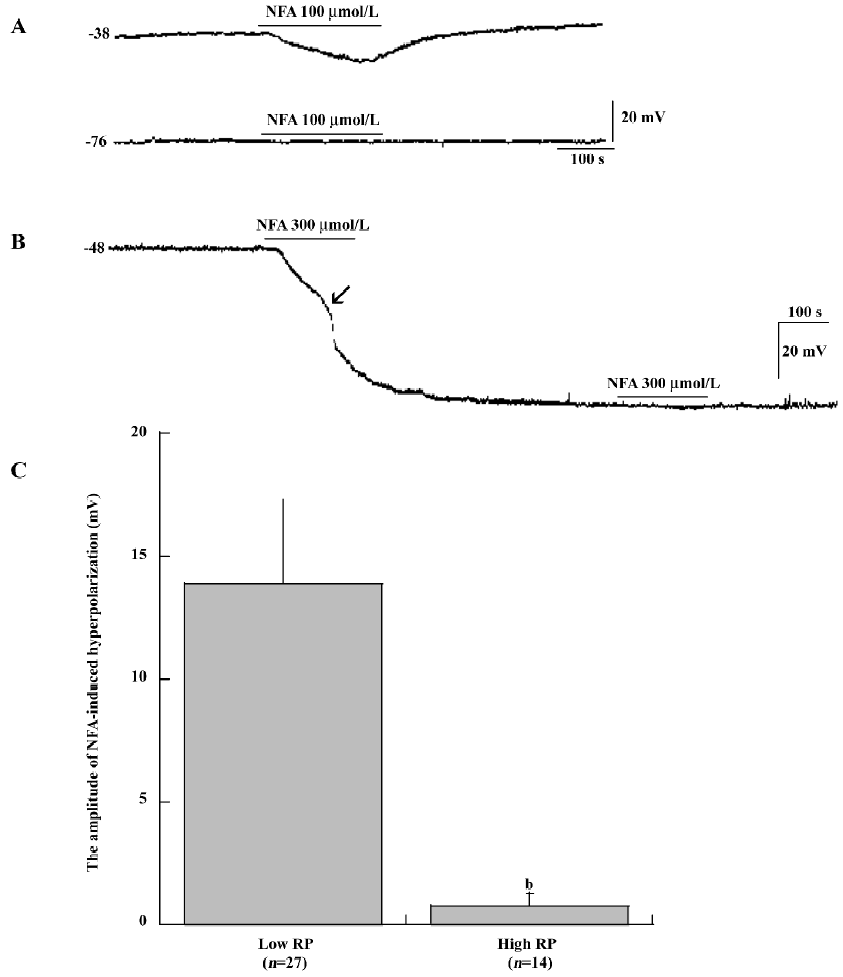
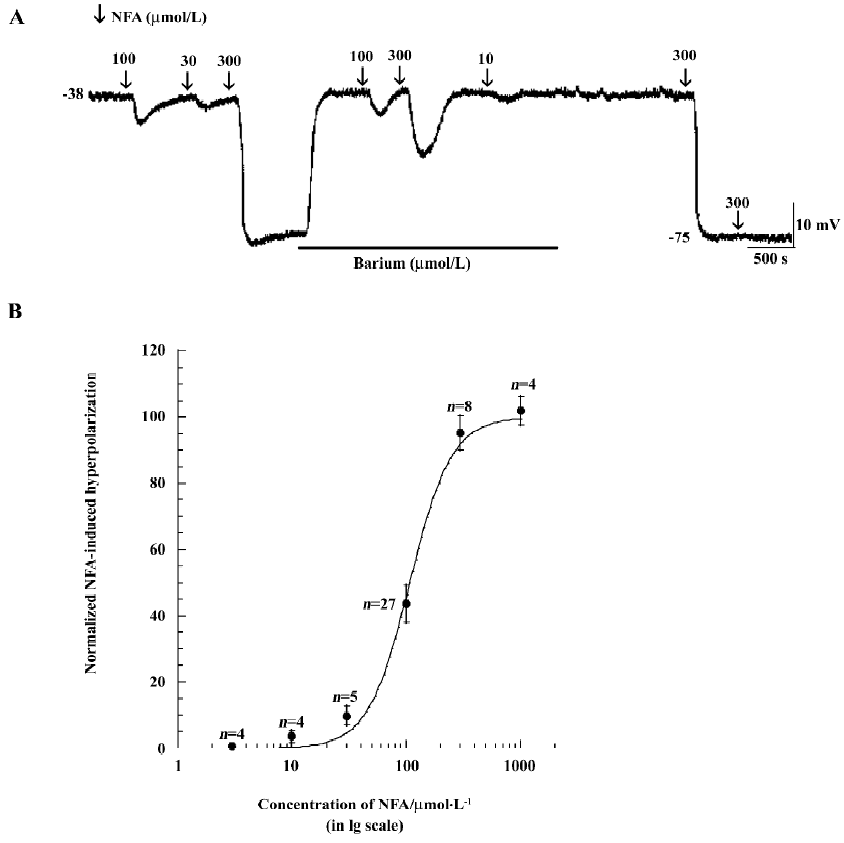

Direct intracellular recordings of SMC membrane potential indicated that membrane potential responses to bath application of the Cl– channel antagonists (10–1000 μmol/L NFA, 0.1–10 μmol/L IAA-94 and 200 μmol/L DIDS) made striking difference between the low RP and high RP types of SMC (Figure 1,2A,4A). In low RP SMC, NFA (100 μmol/L) caused a hyperpolarization of 13.9±3.4 mV (mean±SEM, n=27, P<0.01) from an initial low resting membrane potential of –43.59±1.47 mV (Figure 1A,1C). The amplitude of NFA (100 μmol/L)-induced hyperpolarization ranged from 9.8 mV to 21.6 mV. The NFA-induced responses were concentration -dependent. Figure 2A shows the records of membrane hyperpolarization in response to different concentrations of NFA (10-300 μmol/L) obtained from 1 cell. Figure 2B reveals the concentration–response curve of NFA (3–1000 μmol/L)-induced hyperpolarization. The curve was a good fit for the data to the logistic equation Y=Emax/(1+[Kd/C]n) (where C was the concentration of NFA, and Kd, the dissociation constant of NFA, was 108 μmol/L). The Hill coefficient (n) was 2.38. (In some data, this figure was reached with the pre-application of 50–100 μmol/L barium.) However, NFA (up to 100 μmol/L) had little effect on the membrane potential of cells that had an initial high RP level (Figure 1A) or that had a high RP shifted from a low RP level (Figure 1B).
NFA shifts membrane potential A shift of low RP SMC to a permanent high RP level (approximately –65 mV to –90 mV) was frequently observed. The hyperpolarization shift was triggered by a 1–3 min application of high extracellular potassium (10 mmol/L), acetylcholine (ACh; 3–10 μmol/L), DPTA–NONOate (10 μmol/L, a nitric oxide donor), pinacidil (≥100 μmol/L, an activator of the ATP-sensitive potassium channel), or an unknown spontaneous reason. In these cases, the washout recovery of the membrane potential was aborted[25,26]. In the present experiment, NFA (≥100 μmol/L), IAA-94 (≥10 μmol/L), and DIDS (≥200 μmol/L) also shifted the low RP cells to the high RP cells with an initial RP at a low RP level, the RP further shifted to a level near –75 mV and stayed at this level during the remaining period of recording from 15 min to 2 h (Figure 1B, 2A, 3A, 4A).
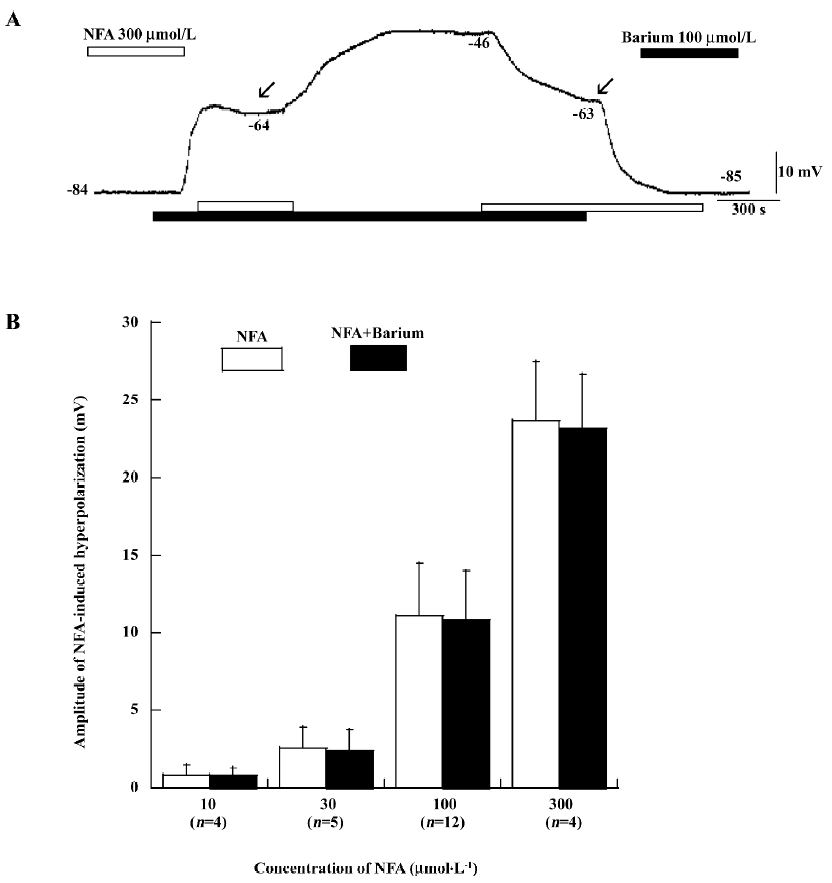
Effects of barium and ouabain on NFA-induced hyperpolarization Barium (1–500 μmol/L) caused a robust depolarization in high RP cells to a low level from –50 to –25 mV [26]. The depolarization induced by 1–50 μmol/L barium was usually completely reversible after a 5–10 min washout. Moreover, in some cells, the RP shifted from a high level (–71 to –85 mV) and remained at a level of approximately -40 mV in the remaining 25–45 min recording time after the 50 μmol/L barium washout. Figure 3A shows that barium (100 μmol/L) could cause depolarization of the high RP cell. Barium (100 μmol/L) and NFA (300 μmol/L) did not shift the RP from a high level to a low level. On the contrary, NFA (300 μmol/L) did not shift the RP from a low level to a high level with simultaneously-incubating barium (100 μmol/L). We noted that NFA (300 μmol/L) maintained the membrane potential around –63 to –64 mV with incubating barium (see arrows of Figure 3A). Ouabain (a Na+–potassium pump current inhibitor, 100 μmol/L)-induced depolarization in the high RP cells was often largely (32.8±1.8 mV, n=10) or fully reversible after 15 min washout. Figure 4A shows that NFA (100 μmol/L) could cause hyperpolarization in low RP cells and shift the membrane potential from a low to high RP level. Ouabain (100 μmol/L) could shift the membrane potential from a high to low RP level again. Moreover, NFA (100 μmol/L) did not shift from a low to high RP level with simultaneously-incubating ouabain (100 μmol/L). Note that the membrane potential of cells automatically shifted from a low to high RP level when ouabain was removed (Figure 4A).
The responses of the cells that had shifted from a high to low RP level with or without barium and ouabain were almost always hyperpolarized by NFA (Figure 2A, 3A, 4A). However, the amplitude of NFA-induced hyperpolarization with simultaneously-incubating barium and ouabain was lower than those seen in the low RP cells without the presence of barium (100 μmol/L) and ouabain (100 μmol/L) (n=7, P>0.05, paired t-test). Figures 3 and 4 reveal the different concentrations of NFA (10–300 μmol/L)-induced hyperpolarization with and without the pre-application of barium (100 μmol/L; Figure 3B) and ouabain (100 μmol/L; Figure 4B).
Moreover, 4-AP (1–10 mmol/L), glipizide (1–10 μmol/L), ChTX (50–100 nmol/L), apamin (50–100 nmol/L), IbTX (100 nmol/L), and TEA (10 mmol/L) also caused either a small depolarization (1–5 mV) or no membrane potential change in high RP cells (n≥4), but never caused a shift to the low RP level. These compounds each caused a 3–10 mV depolarization in the low RP cells (n≥4, data not shown).
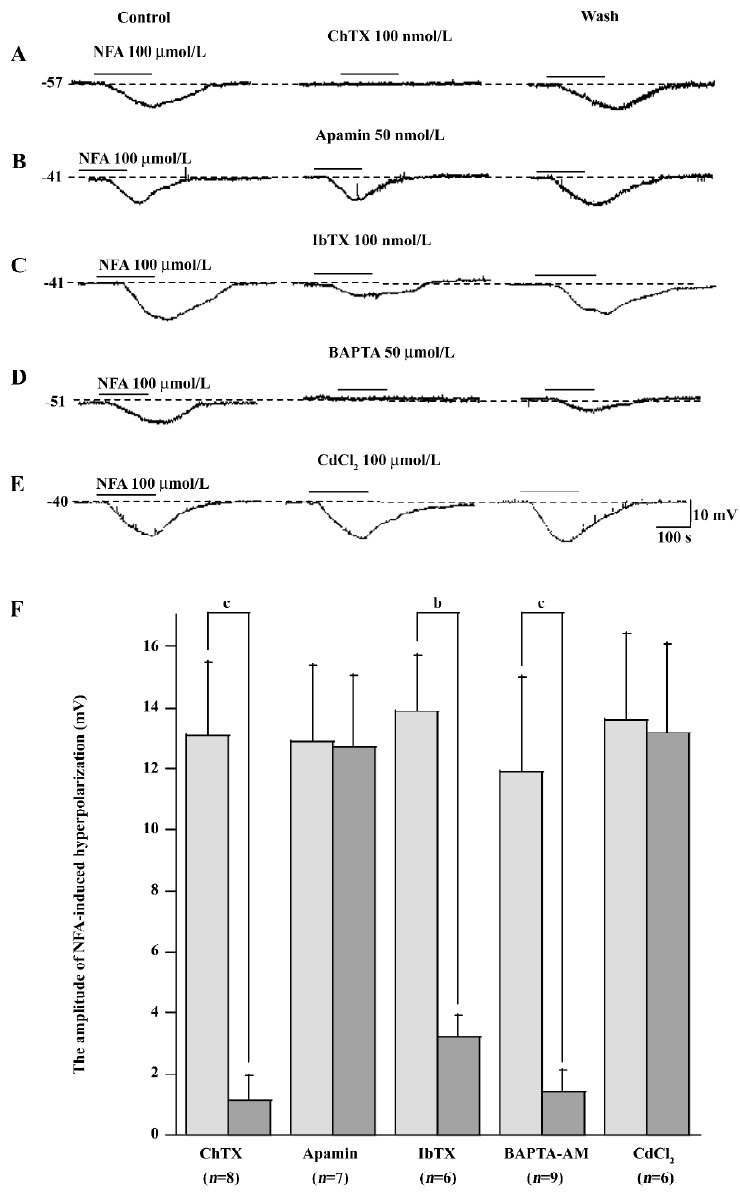
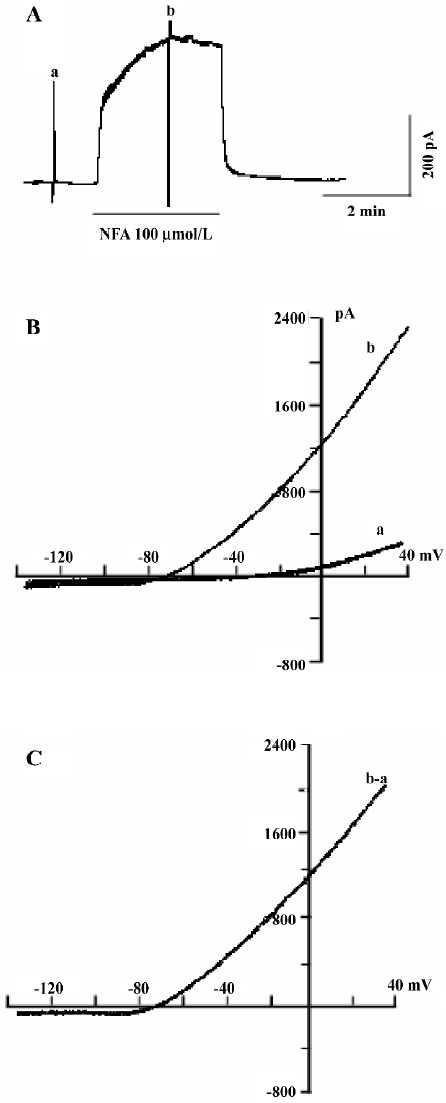
Mechanism of NFA-induced responses To test the mechanism of NFA-induced response in the SMC of the SMA, the effect of antagonists of the potassium channels was obtained. NFA-induced hyperpolarization was almost completely inhibited by IbTX (100 nmol/L, n=6, P<0.05, paired t-test), a specific blocker of large-conductance Ca2+-activated potassium channels (Figure 7C), ChTX (50-100 nmol/L, n=8, P<0.05, paired t-test), a non selective blocker of Ca2+-activated potassium channels (Figure 7A), and TEA (1-10 mmol/L, n=10, P<0.05, paired t-test), a general blocker of a variety of potassium channels, including the Ca2+-activated potassium current (Figure 5A). Besides this, we also used conventional whole-cell patch-clamp to determine what kind of channel was activated by NFA. Figure 6A shows that the gap-free trace represents the trace of the NFA-induced outward current. The 2 deflections (a and b) were whole-cell currents caused by ramp voltage commands applied before (a) and during (b) the NFA-induced outward current. Figure 6B shows the I/V curves (a and b) constructed by the ramp commands (a and b) in Figure 6A. Figure 6C shows the I/V relation of the NFA-induced net current (b–a), which had a reversal potential at –77 mV (after correction for Ra). The reversal potential was very close to the calculated Ek (–83 mV).
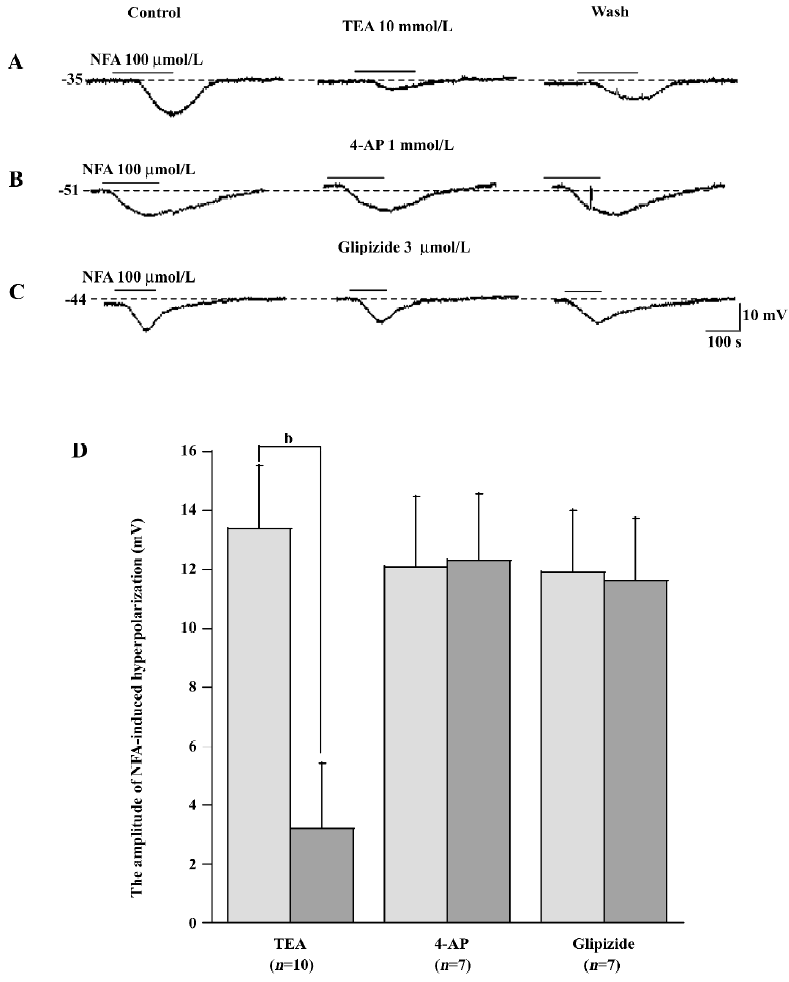
We also studied the effects of 4-AP (a selective voltage-activated potassium channel [KV] blocker), barium (a selective inwardly-rectifying potassium channel [Kir] blocker), glipizide (a selective ATP-sensitive potassium channel [KATP] blocker), apamin (a selective small conductance Ca2+-activated potassium channel [SKCa] blocker) and ouabain (a Na+–potassium pump current inhibitor) on SMC in order to test whether other types of potassium channels take part in the response induced by NFA. However, they had little effect on NFA-induced hyperpolarization in SMC in spite of the concentration of 4-AP (0.5-1 mmol/L, n=7), barium (20-100 μmol/L, n=25), glipizide (3–5 μmol/L, n=7), apamin (50-100 nmol/L, n=7), and ouabain (100 μmol/L, n=19), which were many times the half-block constant when corresponding to potassium channels (Figure 2A,3,4,5B,5C,8)[28,29].
In addition, our experiment found that NFA-induced hyperpolarization could be almost completely inhibited by BAPTA-AM (a membrane-permeant Ca2+ chelator, 50-100 μmol/L, n=9, P<0.05, paired t-test; Figure 7D). After more than 20 min, the response induced by BAPTA-AM largely recovered if the bath time of BAPTA-AM was less than 7 min. However, CdCl2 (a blocker of the non-selective Ca2+ channel, 100 μmol/L, n=6) had little effect on NFA-induced hyperpolarization in SMC (Figure 7E).
The column plots of data statistics exhibited that 100 nmol/L ChTX, 100 nmol/L IbTX, 10 mmol/L TEA, and 50 μmol/L BAPTA-AM had a significant (P<0.05, P<0.01, paired t-test) inhibition, but 1 mmol/L 4-AP, 3 μmol/L glipizide, 50 nmol/L apamin, 10–300 μmol/L barium, 10-300 μmol/L ouabain, and 100 μmol/L CdCl2 had no significant (paired t-test) effects on NFA hyperpolarization.
NFA-induced hyperpolarizations could be blocked by ChTX, IbTX, TEA, and BAPTA-AM, and the reversal potential of the NFA-induced net current was near the Ek. This suggests that the Ca2+-activated potassium channels were involved in the hyperpolarization in SMC.
Discussion
The main findings of this study include: (1) NFA, IAA-94, and DIDS caused concentration-dependent hyperpolarizations in low RP level cells, but not in high RP level cells of the SMA; (2) the NFA-induced hyperpolarization was specifically blocked by a non-selective Ca2+-activated potassium channel blocker (ChTX), a specific large-conductance Ca2+-activated potassium channel blocker (IbTX), a variety of potassium channel general blockers (TEA), and membrane-permeant Ca2+ chelator (BAPTA-AM), where it was not affected by other potassium channel blockers; (3) these triggers (including NFA) had a large membrane hyperpolarization in common, which was a key factor to invoke the maximal Kir and Na+–potassium pump activation and keep it activated in the SMC. Taken together, the results suggest that NFA, a Cl– channel antagonist, hyperpolarizes the vascular cells when the cells are at the low RP level. Our findings extended the observations from jejunum SMC, corneal epithelium cells, and coronary SMC, where NFA and flufenamic acids (a Cl– channel antagonist) increased a calcium-dependent or independent potassium current[23,30-32].
Moreover, the recorded cells were composed of both SMC and EC roughly at a 2:1 ratio (data not shown). Both types of cells showed electrical coupling within the same type of cells and between the 2 types of cells, thus they generally had similar electrical membrane properties; for example, both had 2 RP levels and responded the same way to the application of high potassium and ACh[26], so we refer to both SMC and EC by “SMC” or “cells” unless specified.
Our data support the notion that the NFA-induced hyperpolarization or the outward current in SMC of the SMA is generated by opening the KCa channel. Evidence shows that the hyperpolarization was blocked by ChTX for KCa, IbTX for BKCa[33], BAPTA-AM for chelate intracellular Ca2+, TEA for a variety of potassium channels[34], and the reversal potential of the NFA-induced net current near the Ek, but not by selectively blockers for other potassium channels, including barium for Kir, glipizide for KATP, 4-AP for KV, apamin for SKCa and ouabain for the Na+–potassium pump[12,25,26,35-37]. Multiple types of KCa channels have been described in different systems and in the same tissue, including vascular smooth muscle, and they are differentiated by several parameters, such as conductance, pharmacology, and kinetics[8,38-40]. Large, intermediate, and small conductance KCa channels have all been identified in EC[26,35]. The large-conductance KCa is a special member of the family of ligand-gated potassium channels because its gating is both ligand- and voltage-dependent. Channel opening requires calcium binding to sites on the cytoplasmic face of the channel, and in the presence of calcium, channel opening is increased by membrane depolarization. NFA may activate the BKCa that is sensitive or not to ChTX[23,41]. Ottolia and Toro[23] indicate that BKCa channels possess a specific NFA receptor. Furthermore, the opening of BKCa channels by NFA is caused by an increase in the sensitivity of channel gating to calcium. The NFA binding site is not the same as the one for ChTX and TEA. Generally, the receptor for NFA is not located at or near the pore of the BKCa channel, and NFA association to its receptor does not alter the functional properties of TEA and ChTX receptors located in the external vestibule of the channel pore[23]. Jury et al reported that NFA acts by opening potassium channels, some of them TEA sensitive and some not. BKCa channels are sensitive to both Ca2+ and voltage. It has been postulated that these channels might be involved in the membrane repolarization that follows depolarization and the associated increase in intracellular free calcium during an action potential[8]. Poronnik et al’s work has demonstrated that flufenamic acid can raise intracellular calcium in ST885 cells, a neonatal mouse mandibular line[43]. As the outward potassium current reported in Farrugia et al’s study was not calcium sensitive, changes in intracellular calcium should not be a factor in modulating this current in jejunal SMC[31].
The application of BAPTA-AM indicates that NFA-induced hyperpolarization was dependent on the increase of the intracellular calcium concentration. Recently, it has found that there is overlapping pharmacology of calcium-dependent potassium and calcium-dependent Cl– channels[44]. However, in the present study, NFA-induced hyperpolarization could be blocked by ChTX, IbTX, and TEA. Furthermore, the reversal potential of the NFA-induced outward current was near the Ek (approximately –77 mV; Figure 7D). Also, NFA-induced hyperpolarization was blocked by BAPTA-AM (Figure 8D), not by CdCl2 (Figure 8E). This evidence suggests that the KCa channel is involved in NFA-induced hyperpolarization. However, a lot of questions still remain: for example, where does cytosolic calcium come from, and which receptor can be activated by NFA? This study will be the basis of our significant work in the future.
The elevation of extracellular potassium can activate the Kir and the electrogenic Na+–potassium pump that causes hyperpolarization[45-47]. NFA indirectly hyperpolarizes the SMC through potassium release by activating the KCa, then the increased extracellular potassium activates the Kir and Na+–potassium pump current in SMC. A permanent shift from a low to high RP level can be triggered by high potassium (Kir and Na+–potassium pump activator), ACh[23,26], NFA[23,41] (activating BKCa), pinacidil, and nitric oxide (KATP activators)[25]. Since these triggers (including NFA) have a large membrane hyperpolarization in common, it is suggested that the hyperpolarization itself is a key factor to invoke the maximal Kir and Na+–potassium pump activation and keep it activated.
In summary, using intracellular and tight-seal whole-cell recording methods, we suggest that NFA induces concentration-dependent, reversible hyperpolarization in SMC in the cochlear SMA via the activation of the Ca2+-activated potassium channels. Thus we think NFA is an unreliable pharmacological tool to evaluate Cl- channel contributions to smooth muscle function[42].
References
- Thorne PR, Nuttall AL, Scheibe F, Miller JM. Sound-induced artifact in cochlear blood flow measurements using the laser Doppler flowmeter. Hear Res 1987;31:229-34.
- Ren T, Brown NJ, Zhang M, Nuttall AL, Miller JM. A reversible ischemia model in gerbil cochlea. Hear Res 1995;92:30-7.
- Hultcrantz E. Clinical treatment of vascular inner ear diseases. Am J Otolaryngol 1988;9:317-22.
- Nuttall AL, Hultcrantz E, Larsen HC, Angelborg C. Cochlear blood flow increases after systemic hemodilution: comparison of simultaneous laser Doppler flowmetry and radioactive microsphere measurements. Hear Res 1988;34:215-23.
- Didier A, Miller JM, Nuttall AL. The vascular component of sodium salicylate ototoxicity in the guinea pig. Hear Res 1993;69:199-206.
- Tamhankar M, Solomon D. Acute Hearing Loss. Curr Treat Options Neurol 2004;6:55-65.
- Beech DJ, Bolton TB. Two components of potassium current activated by depolarization of single smooth muscle cells from the rabbit portal vein. J Physiol 1989;418:293-309.
- Latorre R, Oberhauser A, Labarca P, Alvarez O. Varieties of calcium-activated potassium channels. Annu Rev Physiol 1989;51:385-99.
- Kovacs RJ, Nelson MT. ATP-sensitive K+ channels from aortic smooth muscle incorporated into planar lipid bilayers. Am J Physiol 1991;261:H604-9.
- McManus OB. Calcium-activated potassium channels: regulation by calcium. J Bioenerg Biomembr 1991;23:537-60.
- Quayle JM, McCarron JG, Brayden JE, Nelson MT. Inward rectifier K+ currents in smooth muscle cells from rat resistance-sized cerebral arteries. Am J Physiol 1993;265:C1363-70.
- Jiang ZG, Nuttall AL, Zhao H, Dai CF, Guan BC, Si JQ, et al. Electrical coupling and release of K+ from endothelial cells co-mediate ACh-induced smooth muscle hyperpolarization in guinea-pig inner ear artery. J Physiol 2005;564:475-87.
- Crest M, Gola M. Large conductance Ca(2+)-activated K+ channels are involved in both spike shaping and firing regulation in helix neurones. J Physiol 1993;465:265-87.
- Gola M, Crest M. Colocalization of active KCa channels and Ca2+ channels within Ca2+ domains in helix neurons. Neuron 1993;10:689-99.
- Brayden JE, Nelson MT. Regulation of arterial tone by activation of calcium-dependent potassium channels. Science 1992;256:532-5.
- Suarez-Kurtz G, Garcia ML, Kaczorowski GJ. Effects of charybdotoxin and iberiotoxin on the spontaneous motility and tonus of different guinea pig smooth muscle tissues. J Pharmacol Exp Ther 1991;259:439-43.
- Anwer K, Oberti C, Perez GJ, Perez-Reyes N, McDougall JK, Monga M, et al. Calcium-activated K+ channels as modulators of human myometrial contractile activity. Am J Physiol 1993;265:C976-85.
- Khan SA, Mathews WR, Meisheri KD. Role of calcium-activated K+ channels in vasodilation induced by nitroglycerine, acetylcholine and nitric oxide. J Pharmacol Exp Ther 1993;267:1327-35.
- White MM, Aylwin M. Niflumic and flufenamic acids are potent reversible blockers of Ca2(+)-activated Cl– channels in Xenopus oocytes. Mol Pharmacol 1990;37:720-4.
- Korn SJ, Bolden A, Horn R. Control of action potentials and Ca2+ influx by the Ca(2+)-dependent chloride current in mouse pituitary cells. J Physiol 1991;439:423-37.
- McCarty NA, McDonough S, Cohen BN, Riordan JR, Davidson N, Lester HA. Voltage-dependent block of the cystic fibrosis transmembrane conductance regulator Cl– channel by two closely related arylaminobenzoates. J Gen Physiol 1993;102:1-23.
- Greenwood IA, Large WA. Comparison of the effects of fenamates on Ca-activated chloride and potassium currents in rabbit portal vein smooth muscle cells. Br J Pharmacol 1995;116:2939-48.
- Ottolia M, Toro L. Potentiation of large conductance KCa channels by niflumic, flufenamic, and mefenamic acids. Biophys J 1994;67:2272-9.
- Wang YZ, Liu ZJ, Li L, Fan P, Si JQ, Zhao L, et al. Effects of chloride channel blockers on excitatory junction potentials in smooth muscle cells of cochlear spiral modiolar artery in guinea pigs. Sheng Li Xue Bao 2006;58:456-62.
- Si JQ, Zhao H, Yang Y, Jiang ZG, Nuttall AL. Nitric oxide induces hyperpolarization by opening ATP-sensitive K(+) channels in guinea pig spiral modiolar artery. Hear Res 2002;171:167-76.
- Jiang ZG, Si JQ, Lasarev MR, Nuttall AL. Two resting potential levels regulated by the inward-rectifier potassium channel in the guinea-pig spiral modiolar artery. J Physiol 2001;537:829-42.
- Guan BC, Si JQ, Jiang ZG. Blockade of gap junction coupling by glycyrrhetinic acids in guinea pig cochlear artery: a whole-cell voltage- and current-clamp study. Br J Pharmacol 2007;151:1049-60.
- Knot HJ, Zimmermann PA, Nelson MT. Extracellular K(+)-induced hyperpolarizations and dilatations of rat coronary and cerebral arteries involve inward rectifier K(+) channels. J Physiol 1996;492:419-30.
- Robertson BE, Nelson MT. Aminopyridine inhibition and voltage dependence of K+ currents in smooth muscle cells from cerebral arteries. Am J Physiol 1994;267:C1589-97.
- Rae JL, Farrugia G. Whole-cell potassium current in rabbit corneal epithelium activated by fenamates. J Membr Biol 1992;129:81-97.
- Farrugia G, Rae JL, Szurszewski JH. Characterization of an outward potassium current in canine jejunal circular smooth muscle and its activation by fenamates. J Physiol 1993;468:297-310.
- Farrugia G, Rae JL, Sarr MG, Szurszewski JH. Potassium current in circular smooth muscle of human jejunum activated by fenamates. Am J Physiol 1993;265:G873-9.
- Rudy B. Diversity and ubiquity of K channels. Neuroscience 1988;25:729-49.
- Sah P. Ca(2+)-activated K+ currents in neurones: types, physiological roles and modulation. Trends Neurosci 1996;19:150-4.
- Nilius B, Droogmans G. Ion channels and their functional role in vascular endothelium. Physiol Rev 2001;81:1415-59.
- Jiang ZG, Qiu J, Ren T, Nuttall AL. Membrane properties and the excitatory junction potentials in smooth muscle cells of cochlear spiral modiolar artery in guinea pigs. Hear Res 1999;138:171-80.
- Jiang ZG, Shi X, Zhao H, Si JQ, Nuttall AL. Basal nitric oxide production contributes to membrane potential and vasotone regulation of guinea pig in vitro spiral modiolar artery. Hear Res 2004;189:92-100.
- Farley J, Rudy B. Multiple types of voltage-dependent Ca2+-activated K+ channels of large conductance in rat brain synaptosomal membranes. Biophys J 1988;53:919-34.
- Inoue R, Kitamura K, Kuriyama H. Two Ca-dependent K-channels classified by the application of tetraethylammonium distribute to smooth muscle membranes of the rabbit portal vein. Pflugers Arch 1985;405:173-9.
- Reinhart PH, Chung S, Levitan IB. A family of calcium-dependent potassium channels from rat brain. Neuron 1989;2:1031-41.
- Kozak JA, Misler S, Logothetis DE. Characterization of a Ca2+-activated K+ current in insulin-secreting murine betaTC-3 cells. J Physiol 1998;509:355-70.
- Jury J, Patel M, Bowes T, Daniel EE. Actions of putative chloride channel blocking agents on canine lower esophageal sphincter (LES). Can J Physiol Pharmacol 2001;79:1007-14.
- Poronnik P, Ward MC, Cook DI. Intracellular Ca2+ release by flufenamic acid and other blockers of the non-selective cation channel. FEBS Lett 1992;296:245-8.
- Greenwood IA, Leblanc N. Overlapping pharmacology of Ca2+-activated Cl- and K+ channels. Trends Pharmacol Sci 2007;28:1-5.
- Edwards G, Dora KA, Gardener MJ, Garland CJ, Weston AH. K. + is an endothelium-derived hyperpolarizing factor in rat arteries. Nature 1998;396:269-72.
- Weston AH, Richards GR, Burnham MP, Feletou M, Vanhoutte PM, Edwards G. K. +-induced hyperpolarization in rat mesenteric artery: identification, localization and role of Na+/K+-ATPases. Br J Pharmacol 2002;136:918-26.
- Edwards G, Feletou M, Gardener MJ, Thollon C, Vanhoutte PM, Weston AH. Role of gap junctions in the responses to EDHF in rat and guinea-pig small arteries. Br J Pharmacol 1999;128:1788-94.