
Effects of ABCA1 variants on rosiglitazone monotherapy in newly diagnosed type 2 diabetes patients1
Introduction
Type 2 diabetes is a progressive and complex metabolic disorder, which is characterized by chronic hyperglycemia resulting from insulin resistance (IR) and deficiency in insulin secretion[1]. Reduced insulin action in the liver, adipose tissue, and skeletal muscle plays a major role in the pathogenesis of type 2 diabetes[2]. Insulin sensitizer thiazolidinediones (TZDs) are oral hypoglycemic agents acting predominantly by enhancing peripheral insulin sensitivity, reducing glucolipotoxicity, and endogenous insulin secretory demands, and preserving β-cell function[3]. They are agonists of nuclear transcription factor peroxisome proliferator-activated receptor γ (PPARγ) that heterodimerizes with the retinoid X receptor, thereby leading to the transcription of genes involved in glucose and lipid homeostasis[4]. As a new generation of oral antidiabetic drug, TZDs improve insulin sensitivity and reduce glycemia, insulinemia, and dyslipidemia in patients with type 2 diabetes[5].
However, it is reported that the clinical response to TZDs varies. The Prevention of Diabetes study observed that approximately 36% patients did not respond to troglitazone treatment effectively[6]. Wide interindividual variability in drug response may be due to multiple factors, including age, sex, race, accommodation, organ function, placebo effect, clinical stage, and the severity of disease. Genetic polymorphisms in the targets of drug therapy have been increasingly recognized as an important mechanism responsible for the interindividual differences in drug efficacy[7]. Previous pharmacogenetical research of TZDs analyzed single nucleotide polymorphisms (SNPs) on PPARG, ADIPOQ, and CYP2C8[8–14], indicating that genetic variants might be related to variations in the response to TZD treatment.
The ATP-binding cassette transporter subfamily A number 1 (ABCA1) gene is located on chromosome 9q31.1 and is composed of 50 exons extending across a genomic region of 147 kb. The encoded transmembrane protein functions as a transporter of cellular cholesterol and phospholipid to lipid-poor apolipoproteins, especially apolipoprotein A1, which is crucial for high-density lipoprotein biogenesis, as well as the reverse cholesterol transport[15]. Genetic studies have shown that ABCA1 might be a candidate gene of type 2 diabetes. Several genetic variants were associated with diabetes and prediabetic intermediate traits[16,17]. A recent study by Brunham et al[18] further investigated its functional role in β cells. They demonstrated that Abca1 probably had an effect on islet cholesterol homeostasis, subsequently influencing glucose tolerance and insulin secretion. In addition, they found that Abca1 influenced rosiglitazone response in mice. However, whether ABCA1 plays a similar role in humans is yet an unanswered question. Thus, we hypothesized that variants of ABCA1 might contribute to interindividual variation in response to rosiglitazone therapy. In this study, we selected 3 non-synonymous variants of ABCA1, R219K, M883I, and R1587K, to evaluate their effects on rosiglitazone treatment in newly diagnosed patients with type 2 diabetes.
Materials and methods
Patients and study design A total of 105 newly diagnosed patients with type 2 diabetes, defined according to the World Health Organization criteria[19], were derived from the outpatient clinics at 10 hospitals in Shanghai. All patients were naive to prior antidiabetic therapy and treated with rosiglitazone for 48 weeks. We enrolled patients 30–70 years of age, glycated hemoglobin ≥6.5%, and a body mass index (BMI) ≥18.5 kg/m2. For the female patients, post-menopause, surgical sterilization, or effective contraception was required. Excluded criteria were: (i) type 1 diabetes, gestational diabetes, or other specific types; (ii) acute or chronic complications in need of insulin therapy; (iii) significant cardiocerebral, hepatic or nephric disease; (iv) malignant tumor, hematological disease, autoimmune disease, psychiatric disease, or significant digestion and absorption disturbances; (v) current exposure to medication affecting glucose metabolism, such as glucocorticoid; (vi) long-term alcohol or drug abuse; (vii) fasting plasma glucose >13 mmol/L (234 mg/dL) and/or 2 h post-load plasma glucose >18 mmol/L (364 mg/dL); and (viii) blood pressure >180/110 mmHg.
The initial dose was 4 mg/d and escalated to 8 mg/d in patients who failed to attain glycemic targets of fasting plasma glucose >7 mmol/L (126 mg/dL) and/or 2 h plasma glucose >11 mmol/L (200 mg/dL). Patients with glycated hemoglobin was ≥8% or fasting plasma glucose >13 mmol/L (234 mg/dL) or 2 h plasma glucose >18 mmol/L (364 mg/dL) twice (a maximal interval of 6 d) were withdrawn from the study. This study was approved by the institutional review board of Shanghai Jiao Tong University Affiliated Sixth People’s Hospital (Shanghai, China). Each patient provided written informed consent before participating in the study.
Anthropometric measurements General anthropometric parameters, including height (in m), weight (in kg), waist and hip circumferences (in cm), and systolic and diastolic blood pressure (in mmHg) were measured in all patients at baseline and 48 weeks after the initiation of rosiglitazone therapy. BMI and waist–hip ratio were calculated as weight/height2 and waist/hip, respectively.
Clinical laboratory tests Blood samples were collected after an overnight fast and 2 h after a 75 g oral glucose tolerance test (OGTT). Plasma glucose concentrations were measured using the glucose oxidase-peroxidase method with commercial kits (Shanghai Biological Products Institution, Shanghai, China). Glycated hemoglobin values were determined by high-performance liquid chromatography performed on a Bio-Rad Variant II hemoglobin testing system (Bio-Rad Laboratories, Hercules, CA, USA). Serum lipid profiles, including total cholesterol, triglyceride, high-density lipoprotein cholesterol (HDL-C), and low-density lipoprotein cholesterol (LDL-C) were measured with a type 7600-020 Automated analyzer (Hitachi, Tokyo, Japan). An arginine stimulation test was performed to estimate acute insulin secretion of islet β cells. The serum levels of insulin and proinsulin were measured in duplicate at 0, 2, 4, and 6 min (insulin0, insulin2, insulin4, insulin6; proinsulin0, proinsulin2, proinsulin4, proinsulin6) after an intravenous injection of 50 mL arginine solution at the concentration of 10%, using radioimmunoassay (Linco Research, St Charles, MO, USA). The intra-assay coefficients of variation were less than 10%. The evaluation of IR and β-cell secretion at baseline was calculated using the homeostasis model assessment (HOMA) index[20], with the following formula: HOMA–IR=fasting insulin×fasting plasma glucose/22.5, HOMA of β-cell function (HOMA–B)=20×fasting insulin/(fasting plasma glucose–3.5). The amount of acute phase insulin and proinsulin secretion after arginine stimulation was calculated with the following equation: Acute insulin secretion=(insulin2+insulin4+ insulin6)/3–insulin0; acute proinsulin secretion=(proinsulin2+ proinsulin4+proinsulin6)/3–proinsulin0.
Genotyping Genomic DNA was extracted from peripheral blood leucocytes in the whole-blood samples. The SNPs were detected by the PCR restriction fragment length polymorphism. PCR amplification was performed on the GeneAmp PCR system 9700 (Applied Biosystems, Foster City, CA, USA). Amplicons were subsequently digested overnight with restriction enzymes. Following electrophoresis in 12% polyacrylamide gels, the digestion products were stained with ethidium bromide and visualized in the Gel Doc 2000 gel documentation system (Bio-Rad Laboratories, USA). Sixteen random samples were duplicated to confirm the genotyping accuracy, and no discrepancy was detected.
Definition of rosiglitazone responsiveness As there are no generally accepted criteria to divide patients into responders and non-responders, we used 2 methods to define the response to rosiglitazone treatment based on previous clinical studies[11,21–23]. Criterion 1 is a decrease of >15% in all glycemic measures of glycated hemoglobin, fasting, and 2 h plasma glucose after a 75 g glucose OGTT. Criterion 2 is a decrease of 0.5% in glycated hemoglobin. The withdrawals owning to inadequately controlled blood glucose or glycated hemoglobin were defined as non-responders in the analysis.
Statistical analysis Data were shown as mean±SEM or N (%). Allele frequencies were calculated by gene counting. Tests of the Hardy–Weinberg equilibrium were performed[24]. Pairwise linkage disequilibrium was determined by calculating |D´| and r2 using Haploview (version 3.32)[25]. The differences between groups were tested using Student’s t-test or Kruskal–Wallis test when appropriate. Genotype distribution differences between responders and non-responders were compared by Fisher’s exact test or χ2-test. Considering few subjects of rare allele homozygotes, the genotype–phenotype associations were analyzed between common allele homozygotes and rare allele carriers. A 2-tailed P-value ≤0.05 was considered statistically significant. All statistical analyses were performed using SAS for Windows (version 6.12; SAS Institute, Cary, NC, USA).
Results
Clinical characteristics of the patients before and after rosiglitazone treatment Of the 105 patients enrolled, 93 patients (65 men and 28 women, mean age 52.09±9.09 years) completed the entire study. Twelve patients were withdrawn, among whom 5 patients were attributed to inadequately controlled blood glucose or glycated hemoglobin levels; 1 patient had abnormal liver function and 6 patients were lost to follow up.
The baseline and post-therapy clinical characteristics of the study group are summarized in Table 1. After 48 weeks of rosiglitazone therapy, the blood glucose and glycated hemoglobin levels significantly decreased in comparison with baseline (P<0.01). Meanwhile, significant improvements in HOMA–IR (P<0.01), HOMA–B (P<0.01), and acute proinsulin secretion after arginine stimulation (P<0.01) were observed. With regards to the lipid profiles, only the increase in HDL-C level was observed (P<0.01).
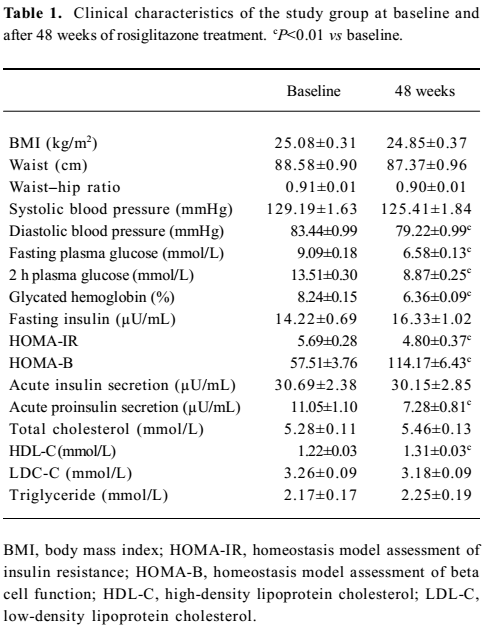
Full table
Association between ABCA1 genetic variants and the response rate of rosiglitazone treatment The genotype distributions of 3 SNPs were consistent with the Hardy–Weinberg equilibrium. By calculating |D´| and r2, a low extent of linkage disequilibrium was detected among them (Table 2). Therefore, the effects of 3 SNPs on rosiglitazone treatment were analyzed, respectively.

Full table
The total response rate of the cohort was 0.398 and 0.782 defined by the 2 criteria, respectively. The genotype frequencies according to the therapeutic responses are shown in Table 3. According to the first criterion, R219K was associated with response to rosiglitazone treatment with more treatment failures in the rare allele homozygotes KK. Eighty-eight percent of the KK homozygotes failed compared with only 52% of the RR homozygotes. The heterozygote RK group showed an intermediate response rate. The per-allele odds ratio for treatment failure was 2.04 (P<0.05). According to the second criterion, although not significant, the same trend was observed; the KK homozygotes had a poor response to rosiglitazone therapy. No significant effect of M883I or R1587K on rosiglitazone therapy was observed.
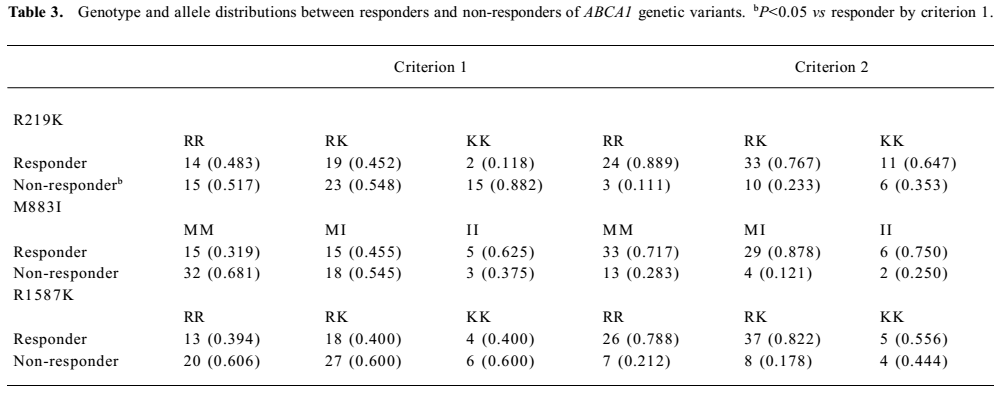
Full table
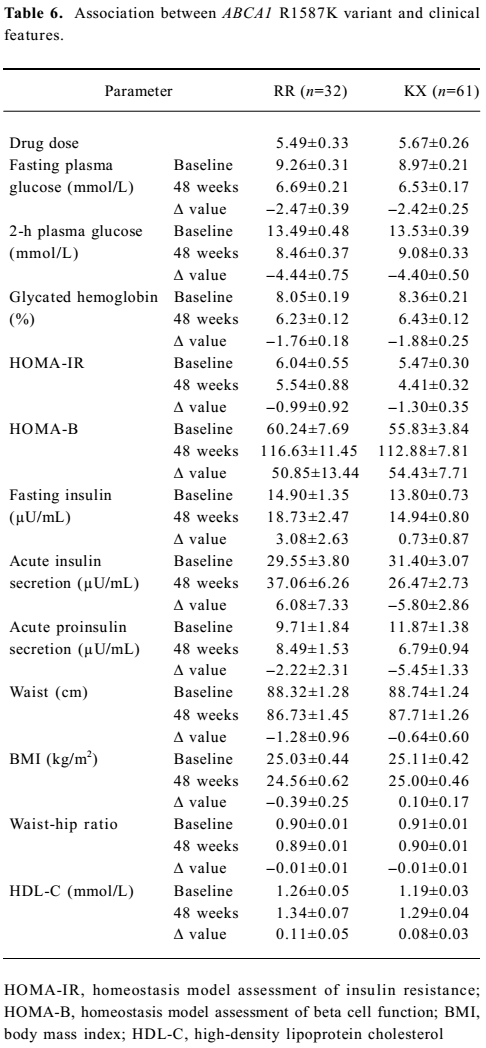
Association between ABCA1 genetic variants and the effect of rosiglitazone treatment on clinical features The association between R219K and clinical features is shown in Table 4. Here we detected a significantly higher 2-h plasma glucose (P<0.05) and waist–hip ratio (P<0.05) at baseline in the RR homozygotes compared with minor K allele carriers. The RR homozygotes also showed greater though non-significant reductions in fasting and 2-h plasma glucose as well as the glycated hemoglobin level after 48 weeks’ treatment. With respect to insulin sensitivity, the decline in the HOMA–IR value was significantly greater in RR homozygotes (P<0.05). As for the lipid profiles, we found no significant differences between the 2 groups. Neither M883I nor R1587K was observed to be associated with clinical features at baseline or after treatment (Tables 5 and 6).
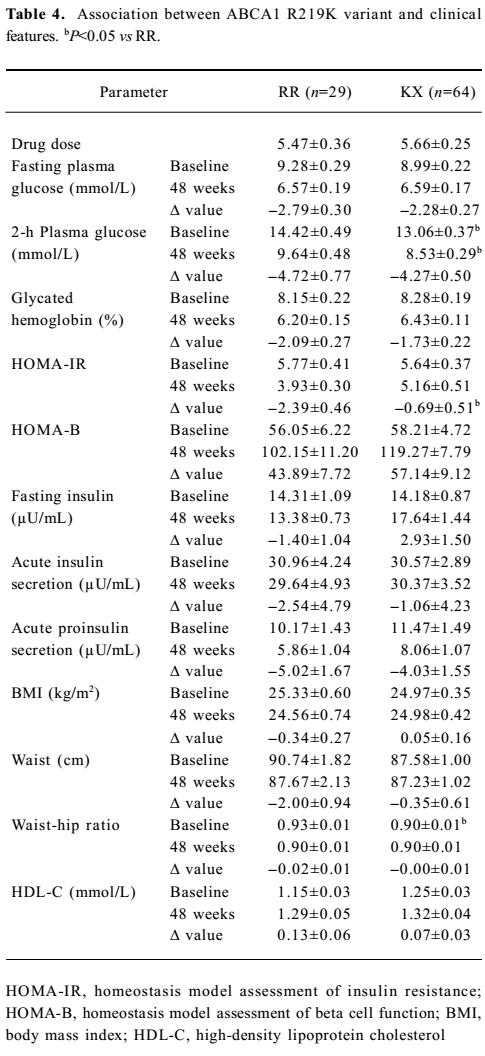
Full table
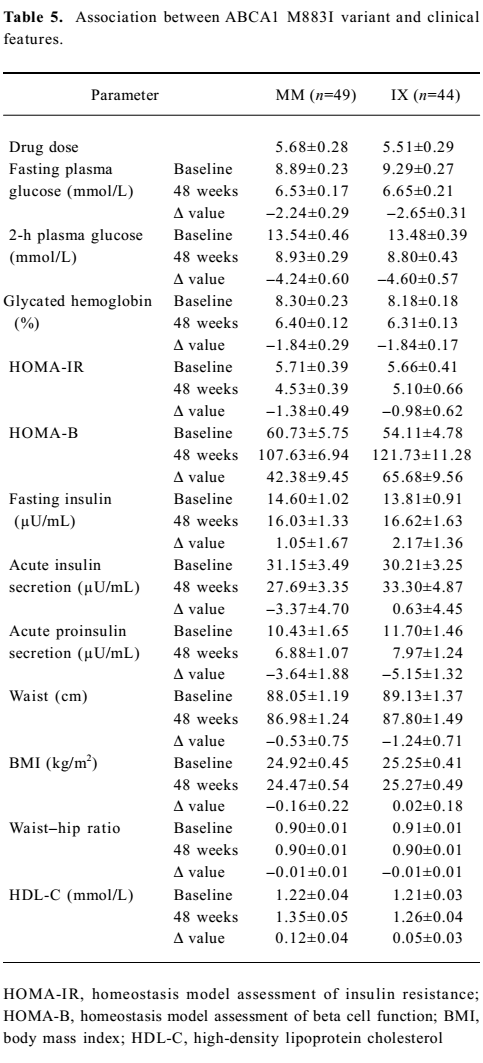
Full table
Full table
Discussion
Pharmacogenetics involve the study of contributions of inherited differences in drug disposition or drug targets to drug response, with the ultimate goal to select the optimal drug therapy and dosages through the use of genetically-guided, individualized treatment[26]. Rosiglitazone, a member of TZDs, is a widely used insulin sensitizer. Data from ADOPT[22] and DREAM[27] showed that initial treatment with rosiglitazone could slow the progressive loss of glycemic control in diabetic patients and reduce diabetes incidences, as well as regress to normoglycemia in individuals with impaired fasting glucose and/or impaired glucose tolerance.
In this study, we found that the R219K variant of the ABCA1 gene was associated with response to rosiglitazone therapy in newly diagnosed type 2 diabetes patients. The RR homozygotes showed a lower failure rate and a better improvement in insulin sensitivity after 48 weeks’ treatment. Rosiglitazone increases the expression of ABCA1 through a PPARγ-LXRα-ABCA1 pathway, thereby regulating cholesterol homeostasis[28,29]. It is well recognized that accumulation of lipids in tissues leads to β-cell dysfunction and IR[30–32]. Brunham et al[18] further demonstrated that Abca1 had an effect on β-cell function through its role on cholesterol accumulation in islets. Their results indicated that Abca1 mainly affected insulin secretion. However, we failed to find any association between ABCA1 genetic variants and acute insulin or proinsulin secretion after arginine stimulation. The main outcome of our study was that R219K was associated with insulin sensitivity improvement. The underlying mechanism is still unknown.
We did not detect significant differences in lipid profiles between the RR homozygotes and minor K allele carriers, although the latter were suggested to be associated with decreased triglyceride and a trend toward increased HDL-C in some[33,34], but not all[35] association studies. As serum cholesterol levels are influenced by many factors, we suppose that a small change in ABCA1 activity could affect rosiglitazone response without markedly impacting circulating cholesterol profiles.
There are several limitations of this study that should be noted. First, the sample size of this study is relatively small, thus we do not have enough statistical power to detect effects of genetic variants and HDL-C levels. Although previous studies reported that M883I had an impact on ABCA1 function[36] and R1587K was associated with HDL-C level[33,37], we failed to detect any effect of these 2 variants on rosiglitazone treatment. We cannot exclude the possibility that the sample size may be one of the reasons. Second, as most variables of our study are in skew distribution, the Kruskal–Wallis test was the main method used to analyze the association between genotypes and phenotypes, thus we could not adjust for the effect of confounding factors, such as drug dose. However, we did not detect a significant difference in drug dose between groups. The K allele carriers of the R219K variant showed a relatively higher mean drug dose compared with the RR homozygotes, which may also reflect a poorer response to rosiglitazone treatment. Third, a 48 week follow-up period might be inadequate to see the thorough therapeutic effect, but as documented in the ADOPT study[22], the maximal treatment effect of rosiglitazone on glycated hemoglobin was achieved within one year. Considering the compliance of patients, the effect of genotypes on long-term drug response would be better studied in a carefully controlled clinical trial.
In conclusion, we provide evidence that the R219K variant of the ABCA1 gene either directly or as a marker with additional functional variant in linkage disequilibrium, has an effect on response to rosiglitazone treatment. Never-theless, we are at an early stage of defining pharmacogenetic determinants of rosiglitazone treatment response. Long-term follow-up studies with large samples are needed to further confirm our findings and allow individualizing therapy based on genomic information.
Acknowledgements
We thank all the patients taking part in this research. We are grateful to the doctors and nurses who participated in this study from Shanghai Jiao Tong University Affiliated Sixth People’s Hospital; First People’s Hospital; Renji Hospital; Ruijin Hospital, Xinhua Hospital, Fudan University Affiliated Zhongshan Hospital; Huashan Hospital, Second Military Medical University Affiliated Changzheng Hospital; Changhai Hospital, and Tongji University Affiliated East Hospital.
References
- Stumvoll M, Goldstein BJ, van Haeften TW. Type 2 diabetes: principles of pathogenesis and therapy. Lancet 2005;365:1333-46.
- DeFronzo RA. Pathogenesis of type 2 (non-insulin dependent) diabetes mellitus: a balanced overview. Diabetologia 1992;35:389-97.
- Elte JW, Blickle JF. Thiazolidinediones for the treatment of type 2 diabetes. Eur J Intern Med 2007;18:18-25.
- Rosen ED, Spiegelman BM. PPARγ: a nuclear regulator of metabolism, differentiation, and cell growth. J Biol Chem 2001; 276: 37 731–4.
- Day C. Thiazolidinediones: a new class of antidiabetic drugs. Diabet Med 1999;16:179-92.
- Buchanan TA, Xiang AH, Peters RK, Kjos SL, Marroquin A, Goico J, et al. Preservation of pancreatic beta-cell function and prevention of type 2 diabetes by pharmacological treatment of insulin resistance in high-risk Hispanic women. Diabetes 2002;51:2796-803.
- Eichelbaum M, Ingelman-Sundberg M, Evans WE. Pharmaco-genomics and individualized drug therapy. Annu Rev Med 2006;57:119-37.
- Kirchheiner J, Thomas S, Bauer S, Tomalik-Scharte D, Hering U, Doroshyenko O, et al. Pharmacokinetics and pharmacodynamics of rosiglitazone in relation to CYP2C8 genotype. Clin Pharmacol Ther 2006;80:657-67.
- Kang ES, Park SY, Kim HJ, Ahn CW, Nam M, Cha BS, et al. The influence of adiponectin gene polymorphism on the rosiglitazone response in patients with type 2 diabetes. Diabetes Care 2005;28:1139-44.
- Snitker S, Watanabe RM, Ani I, Xiang AH, Marroquin A, Ochoa C, et al. Changes in insulin sensitivity in response to troglitazone do not differ between subjects with and without the common, functional Pro12Ala peroxisome proliferator-activated receptor-γ2 gene variant: results from the troglitazone in Prevention of Diabetes (TRIPOD) study. Diabetes Care 2004;27:1365-8.
- Bluher M, Lubben G, Paschke R. Analysis of the relationship between the Pro12Ala variant in the PPAR-gamma2 gene and the response rate to therapy with pioglitazone in patients with type 2 diabetes. Diabetes Care 2003;26:825-31.
- Wolford JK, Yeatts KA, Dhanjal SK, Black MH, Xiang AH, Buchanan TA, et al. Sequence variation in PPARG may underlie differential response to troglitazone. Diabetes 2005;54:3319-25.
- Kang ES, Park SY, Kim HJ, Kim CS, Ahn CW, Cha BS, et al. Effects of Pro12Ala polymorphism of peroxisome proliferator-activated receptor γ2 gene on rosiglitazone response in type 2 diabetes. Clin Pharmacol Ther 2005;78:202-8.
- Florez JC, Jablonski KA, Sun MW, Bayley N, Kahn SE, Shamoon H, et al. Effects of the type 2 diabetes-associated PPARG P12A polymorphism on progression to diabetes and response to troglitazone. J Clin Endocrinol Metab 2007;92:1502-9.
- Attie AD. ABCA1: at the nexus of cholesterol, HDL and atherosclerosis. Trends Biochem Sci 2007;32:172-9.
- Daimon M, Ji G, Saitoh T, Oizumi T, Tominaga M, Nakamura T, et al. Large-scale search of SNPs for type 2 DM susceptibility genes in a Japanese population. Biochem Biophys Res Commun 2003;302:751-8.
- Daimon M, Kido T, Baba M, Oizumi T, Jimbu Y, Kameda W, et al. Association of the ABCA1 gene polymorphisms with type 2 DM in a Japanese population. Biochem Biophys Res Commun 2005;329:205-10.
- Brunham LR, Kruit JK, Pape TD, Timmins JM, Reuwer AQ, Vasanji Z, et al. Beta-cell ABCA1 influences insulin secretion, glucose homeostasis and response to thiazolidinedione treatment. Nat Med 2007;13:340-7.
- Alberti KG, Zimmet PZ. Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabet Med 1998;15:539-53.
- Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985;28:412-9.
- Scheen AJ, Lefebvre PJ. Troglitazone: antihyperglycemic activity and potential role in the treatment of type 2 diabetes. Diabetes Care 1999;22:1568-77.
- Kahn SE, Haffner SM, Heise MA, Herman WH, Holman RR, Jones NP, et al. Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N Engl J Med 2006;355:2427-43.
- Rosenstock J, Baron MA, Dejager S, Mills D, Schweizer A. Comparison of vildagliptin and rosiglitazone monotherapy in patients with type 2 diabetes: a 24-week, double-blind, randomized trial. Diabetes Care 2007;30:217-23.
- Wigginton JE, Cutler DJ, Abecasis GR. A note on exact tests of Hardy-Weinberg equilibrium. Am J Hum Genet 2005;76:887-93.
- Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 2005;21:263-5.
- Weinshilboum RM, Wang L. Pharmacogenetics and pharmaco-genomics: development, science, and translation. Annu Rev Genomics Hum Genet 2006;7:223-45.
- Gerstein HC, Yusuf S, Bosch J, Pogue J, Sheridan P, Dinccag N, et al. Effect of rosiglitazone on the frequency of diabetes in patients with impaired glucose tolerance or impaired fasting glucose: a randomised controlled trial. Lancet 2006;368:1096-105.
- Chawla A, Boisvert WA, Lee CH, Laffitte BA, Barak Y, Joseph SB, et al. A PPARγ–LXR–ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol Cell 2001;7:161-71.
- Chinetti G, Lestavel S, Bocher V, Remaley AT, Neve B, Torra IP, et al. PPAR-alpha and PPAR-gamma activators induce cholesterol removal from human macrophage foam cells through stimulation of the ABCA1 pathway. Nat Med 2001;7:53-8.
- Unger RH. Lipotoxicity in the pathogenesis of obesity-dependent NIDDM. Genetic and clinical implications. Diabetes 1995;44:863-70.
- Robertson RP, Harmon J, Tran PO, Poitout V. Beta-cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes 2004;53 Suppl 1:S119-24.
- Poitout V, Robertson RP. Mini review: secondary beta-cell failure in type 2 diabetes—a convergence of glucotoxicity and lipotoxicity. Endocrinology 2002;143:339-42.
- Clee SM, Zwinderman AH, Engert JC, Zwarts KY, Molhuizen HO, Roomp K, et al. Common genetic variation in ABCA1 is associated with altered lipoprotein levels and a modified risk for coronary artery disease. Circulation 2001;103:1198-205.
- Soro-Paavonen A, Naukkarinen J, Lee-Rueckert M, Watanabe H, Rantala E, Soderlund S, et al. Common ABCA1 variants, HDL levels, and cellular cholesterol efflux in subjects with familial low HDL. J Lipid Res 2007;48:1409-16.
- Evans D, Beil FU. The association of the R219K polymorphism in the ATP-binding cassette transporter 1 (ABCA1) gene with coronary heart disease and hyperlipidaemia. J Mol Med 2003;81:264-70.
- Brunham LR, Singaraja RR, Pape TD, Kejariwal A, Thomas PD, Hayden MR. Accurate prediction of the functional significance of single nucleotide polymorphisms and mutations in the ABCA1 gene. PLoS Genet 2005;1:e83.
- Frikke-Schmidt R, Nordestgaard BG, Jensen GB, Tybjaerg-Hansen A. Genetic variation in ABC transporter A1 contributes to HDL cholesterol in the general population. J Clin Invest 2004;114:1343-53.