
Modulation of synaptic GABAA receptor function by zolpidem in substantia nigra pars reticulata1
Introduction
The substantia nigra pars reticulata (SNr) occupies one of the output centers of the basal ganglia circuit. It is well known that the inhibitory neurotransmitter γ-aminobutyric acid (GABA) is the major neurotransmitter used in the SNr, which suggests that GABA-mediated neurotransmission in this nucleus plays an important function[1]. Anatomical and electrophysiological evidence has shown that the major sources of GABAergic inputs in the SNr are the striatum, globus pallidus, and local axon collaterals of GABAergic output neurons[2–5].
Moderate to high densities of GABAA receptors were expressed in the SNr[6,7], which mediate fast inhibitory synaptic transmission. GABAA receptors are heteropentameric structures assembled from various subunits, including α1–6, β1–4, γ1–3, δ, ε, π, θ, and ρ1–3. The subunit combination of a particular GABAA receptor plays a crucial role in determining its pharmacological properties and physiological functions. Most GABAA receptors contain at least both α and β subunits, with 1 or more of the other subunits. There are several binding sites within GABAA receptors that interact with a diverse rage of compounds[8]. The benzodiazepine binding site is one of the potent modulating sites, which leads to anxiolytic, anticonvulsant, and sedative effects by potentiating GABA currents. Zolpidem is an imidazopyridine agonist with a high affinity on the benzodiazepine site containing the α1 subunit. Early studies indicated that the highest level of zolpidem binding sites occurred in the SNr[9]. Quantitative autoradiography has shown that 6-hydroxydopamine lesion significantly increased the binding of zolpidem in SNr, which may reflect a compensatory alteration[10,11]. The systemic administration of zolpidem produced an inhibition on the firing of the SNr neurons[12]. The microinfusion of zolpidem and other benzodiazepine agonists into the SNr produced anticonvulsant effects on clonic and tonicclonic seizures[13,14]. Recently, a few clinic reports suggested that zolpidem exerted a therapeutic effect on some groups of Parkinson’s patients[15–17]. However, the direct pharmacological effects of zolpidem in the SNr are not known. In the present study, whole-cell patch-clamp recordings and intranigral microinjection were used to study the function of zolpidem in the rat SNr.
Materials and methods
In vitro slice preparation Sprague-Dawley rats aged 12–14 d were used for the preparation of acute brain slices. The animals were killed by decapitation. The brains were then immediately removed and placed in ice-cold artificial cerebrospinal fluid (ACSF) of the following composition (in mmol/L): NaCl 125, KCl 2.0, MgSO4 1.2, CaCl2 2.5, KH2PO4 1.2, glucose 11, and NaHCO3 26, which was continuously bubbled with 95% O2 and 5% CO2. Midbrain coronal slices (250 µm) containing the SNr were sectioned using a vibrating microtome (Camden Instruments, Loughborough, UK) as previously described[18]. Briefly, after equilibration for at least 1 h, the slices were transferred to a small volume chamber mounted on an upright microscope (Zeiss Axioskop, Oberkochen, Germany) and superfused with ACSF at a rate of 1.5–2.0 mL/min maintained at a temperature of 34 °C. Neuronal soma of the SNr neurons was directly visualized by a combination of differential interference contrast optics and contrast-enhanced infrared video microscopy.
Patch-clamp recording Whole-cell patch-clamp recordings from the SNr neurons were obtained using a patch-clamp amplifier (LM/PCA, List Medical, Darmstadt, Germany). Whole-cell pipettes typically had a resistance of 3–4 MΩ when filled with an internal solution of the following composition (in mmol/L): KCl 140, EGTA 1.0, MgCl2 2.0, Na2ATP 2.0, Tris GTP 0.4, and HEPES 10; the pH was adjusted to 7.25–7.30 with 1 mol/L KOH. The inclusion of 140 mmol/L KCl in the recording pipettes reversed the polarity of GABAA receptor-mediated currents from outward to inward. Monitoring through a camera, a pipette was placed on the soma of a SNr neuron and whole-cell recording was made. Normally, no series resistance compensation was applied, but the cell was rejected if the series resistance increased significantly (>20%) during recording. (F)-2-Amino-5-phosphonopen-tanoic acid (AP5; 50 µmol/L) and 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; 20 µmol/L) were included to eliminate glutamate receptor-mediated synaptic currents. Miniature postsynaptic currents (mPSC) were isolated by the addition of 0.5 µmol/L tetrodotoxin (TTX) to the ACSF. Inhibitory postsynaptic currents were recorded at a holding potential of -70 mV. The current signal was filtered at 3 kHz. Online or offline digitization (10 kHz) was made via the Digidata-pClamp system (Axon Instruments, California, USA). Computer files containing information of synaptic currents were analyzed by the mini analysis program (version 6, Synaptosoft, Decatur, GA, USA), which automatically generates various parameters, including the time of occurrence, peak amplitude, and kinetics.
In vivo surgery Sprague-Dawley rats (250–280 g) were used. The animals were individually housed in a temperature regulated room and maintained on a 12 h:12 h light/dark cycle. All rats had free access to food and water. On the day of surgery, the rats were anesthetized with chloral hydrate (400 mg/kg, intraperitoneally) and placed in a stereotaxic frame. A guide cannula constructed from stainless steel (outer diameter: 0.4 mm; inner diameter: 0.3 mm) was implanted into the SNr (5.5 mm posterior, 2.3 mm lateral from the bregma, 8.0 mm ventral from the skull surface) on either side. The cannulae were fixed to the skull with stainless steel screws and dental acrylic. Stainless steel stylets were used to keep the cannulae sealed.
Rotational test Following at least 3 d of recovery, the rats were tested for motor/turning behavior. Drugs (0.2 µL) were microinjected into the SNr in awake animals over a 2 min period. Injection cannulae were connected to a 1.0 μL microsyringe and a drug was injected. At the end of injection, the cannula was left in the SNr for an additional 1 min before removal and then replaced by a stylet. The net rotational behavior was calculated for 30 min.
Histological controls After the test, the rats were perfused with 10% formalin solution transcardially. Fifty micrometer brain slices were cut to examine the sites of injection. Only the data obtained from animals with correct placement of the cannulae were used for the analysis.
Drugs and statistics Zolpidem and flumazenil were purchased from Tocris (Avonmouth, UK). AP5, CNQX, bicuculline, and TTX were obtained from Sigma (St Louis, MO, USA). The data are expressed as mean±SEM. The Kolmogorov-Smirnov test was used to compare 2 distributions of synaptic current inter-event intervals, amplitudes, decay time, and rise time setting the threshold of significance at a probability (P) of 0.05. Otherwise, paired or unpaired Student’s t-test was applied using a value of 0.05. The net rotational behavior was analyzed by the non-parametric one-way Kruskal-Wallis test followed by the Mann–Whitney U-test.
Results
Identification of GABA and dopamine neurons It is known that 2 types of neurons, GABA and dopamine neurons, exist in the SNr. In the present study, the distinction between dopamine and GABA neurons was based on their electrophysiological characteristics[19,20]. Briefly, dopaminergic neurons displayed a low resting firing rate, slow hyperpolarization after each action potential, and inward rectification in response to hyperpolarizing current injection. In contrast, GABAergic neurons were characterized by a relatively high-frequency firing and absence of the inward rectification. The neurons obtained in the present study were medium-sized neurons, which exhibited the electrophysiological characteristics of GABAergic neurons.
Effects of 100 nmol/L zolpidem on mPSC The mPSC were found in all the SNr neurons, which were sensitive to the GABAA receptor antagonist bicuculline (10 µmol/L). As shown in Figure 1, superfusion of 100 nmol/L zolpidem significantly prolonged the decay time, the time required for the amplitude of the mPSC to decay to half its peak value, without any change in the amplitude, rise time, and inter-event intervals. In 6 cells recorded, 100 nmol/L zolpidem significantly prolonged the decay time (control: 7.4±0.5 ms; zolpidem: 10.3±0.3 ms, P<0.01). The prolongation on decay time was only partially reversed after 20 min of washing. No significant change was observed in amplitude (control: 64.7±5.8 pA; zolpidem: 70.5±7.6 pA, P>0.05), rise time (control: 3.0±0.3 ms; zolpidem: 3.1±0.2 ms, P>0.05) and frequency (control: 2.9±0.6 Hz; zolpidem: 3.0±0.6 Hz, P>0.05).
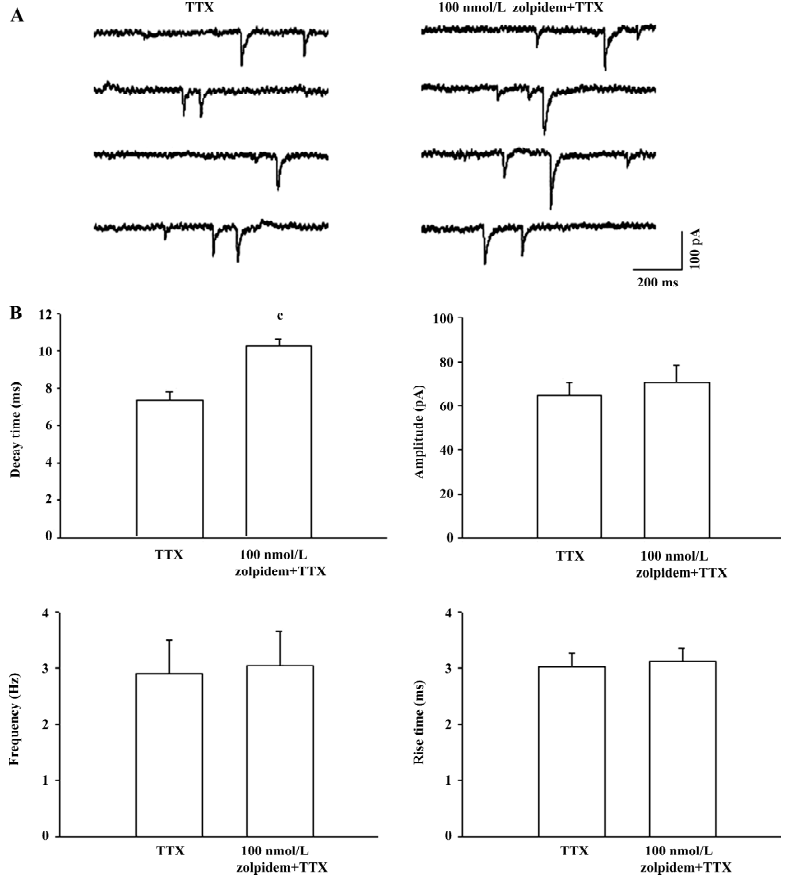
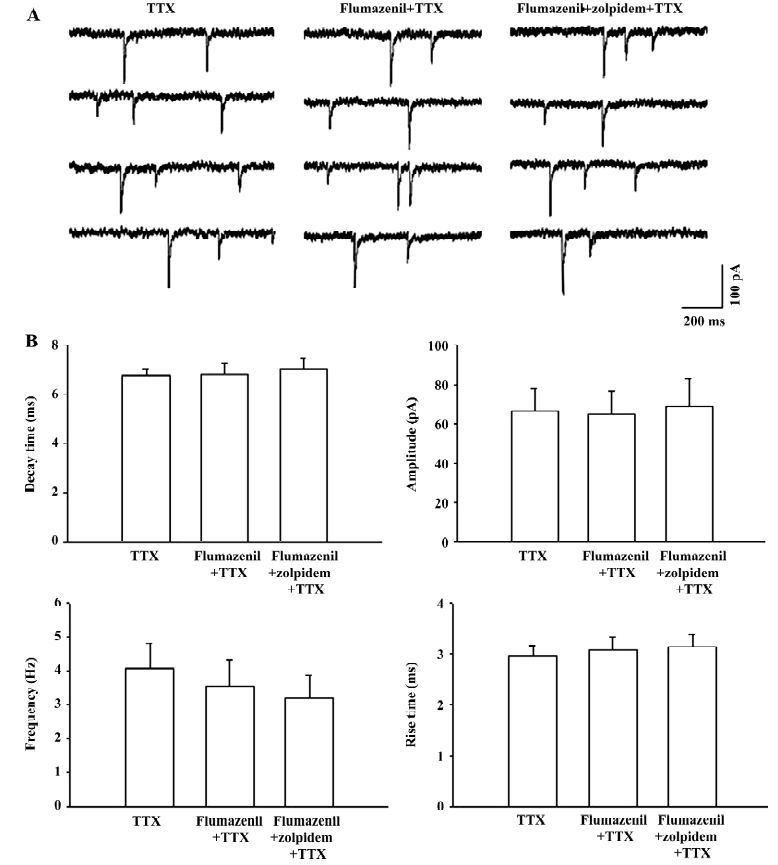
Benzodiazepine antagonist blocked the zolpidem-mediated prolongation of mPSC A further experiment was performed to investigate the effects of the benzodiazepine binding site antagonist flumazenil on the zolpidem-mediated prolongation of the decay time. As shown in Figure 2, 10 µmol/L flumazenil alone did not induce any change on decay time (control: 6.8±0.3 ms; flumazenil: 6.9±0.4 ms, n=8, P>0.05), amplitude (control: 66.7±11.2 pA; flumazenil: 65.2±11.6 pA, P>0.05), frequency (control: 4.1±0.7 Hz; flumazenil: 3.5±0.8 Hz, P>0.05), and rise time (control: 2.9±0.2 ms; flumazenil: 3.1±0.3 ms, P>0.05) of mPSC. However, in the presence of flumazenil, the zolpidem-induced prolongation of the decay time could be completely prevented (flumazenil+zolpidem: 7.1±0.4 ms, P>0.05 compared with flumazenil alone). No significant difference was observed for amplitude (flumazenil+zolpidem: 68.8±13.9 pA, P>0.05), frequency (flumazenil+zolpidem: 3.2±0.7 Hz, P>0.05), and rise time (flumazenil+zolpidem: 3.1±0.2 ms, P>0.05) after flumazenil and zolpidem cosuperfusion.
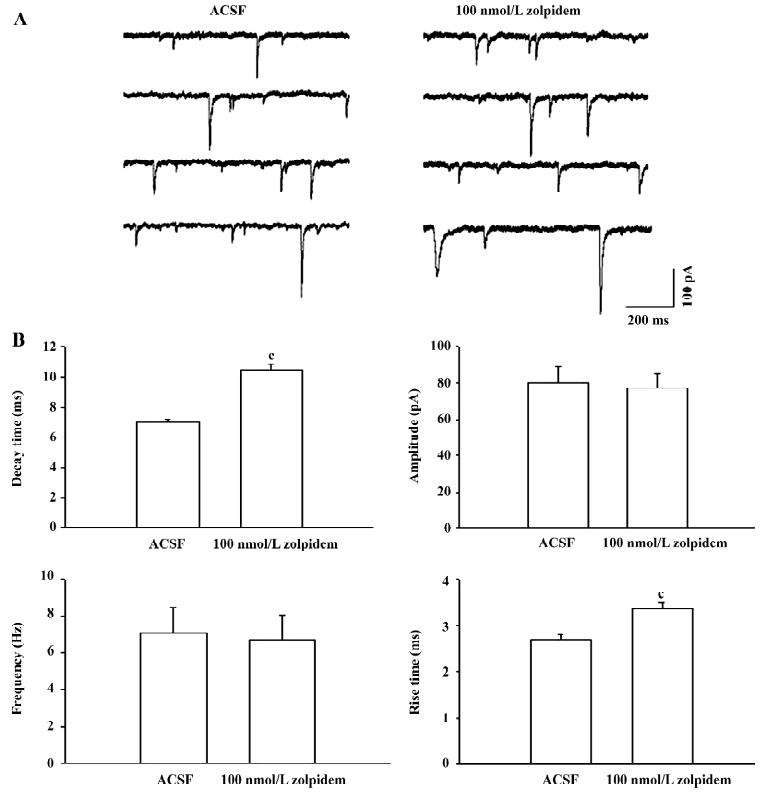
Effects of 100 nmol/L zolpidem on spontaneous PSC The effects of zolpidem on spontaneous PSC (sPSC) were tested. As shown by the example in Figure 3A, 100 nmol/L zolpidem prolonged the decay time and rise time of sPSC significantly. In the 7 neurons studied, zolpidem prolonged the decay time from 7.0±0.2 ms to 10.5±0.4 ms (P<0.01) and the rise time from 2.7±0.1 ms to 3.4±0.1 ms (P<0.01). There was no significant effect on amplitude (control: 80.1±8.8 pA; zolpidem:77.0±8.0 pA, P>0.05) and frequency (control: 7.1±1.4 Hz; zolpidem: 6.7±1.3 Hz, P>0.05). Figure 3B summarizes the effects of 100 nmol/L zolpidem on sPSC. The increase on the decay time of sPSC (49.9%±5.4%) was similar to that of mPSC (40.7%±4.4%, P>0.05).
Effects of 1 µmol/L zolpidem on mPSC and sPSC Further studies were performed to observe the effects of a higher concentration of zolpidem on both mPSC and sPSC. In 6 neurons studied, 1 µmol/L zolpidem prolonged the decay time of mPSC from 6.2±0.4 ms to 11.6±0.6 ms (P<0.01) and amplitude from 65.5±5.9 pA to 77.1±7.2 pA (P<0.05). There was no significant change in the rise time (control: 2.5±0.3 ms; zolpidem: 3.0±0.2 ms, P>0.05) and frequency (control: 3.8±0.7 Hz; zolpidem: 3.7±0.4 Hz, P>0.05). The increase in the decay time induced by 1 µmol/L zolpidem was 87.8%±8.1%, which was significantly stronger than that induced by the concentration of 100 nmol/L (40.7%±4.4%, P<0.05, Figure 4A).
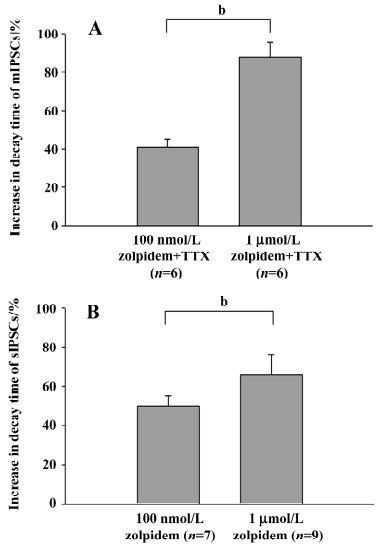
In another 9 neurons studied, 1 µmol/L zolpidem prolonged the decay time of sPSC from 6.9±0.3 ms to 11.3±0.5 ms (P<0.01) and the rise time from 2.5±0.1 ms to 3.0±0.1 ms (P<0.01). There was no significant change in amplitude (control: 67.1±9.3 pA; zolpidem: 79.1±7.9 pA, P>0.05) and frequency (control: 6.5±1.1 Hz; zolpidem: 6.2±1.0 Hz, P>0.05). The increase in the decay time induced by 1 µmol/L zolpidem was 65.6%±10.4%, which was significantly stronger than that induced by 100 nmol/L zolpidem (49.9%±5.4%, P<0.05, Figure 4B). However, no difference was observed between the prolongation of the decay time of mPSC (87.8%± 8.1%) and sPSC (65.6%±10.4%, P>0.05).
Effects of zolpidem on rotational behavior To determine the modulation of zolpidem on motor behavior in vivo, zolpidem was microinjected into the SNr. In 7 rats tested, a unilateral microinjection of zolpidem (1 mmol/L, 0.2 µL) induced a robust contralateral rotation (32.7±2.9 turns/30 min). However, the vehicle microinjection only induced 0.7±0.3 turns/30 min contralateral rotation (n=6, P<0.01 compared with zolpidem). In another 5 rats, a unilateral microinjection of flumazenil (3 mmol/L, 0.2 µL) alone did not produce any significant rotation (2.2±2.1 turns/30 min contralateral rotation, P>0.05 compared with the control). However, the presence of flumazenil could completely block the contralateral rotation induced by the zolpidem microinjection (2.9±2.7 turns/30 min contralateral rotation, n=7, P<0.05 compared with zolpidem alone). These data are summarized in Figure 5.
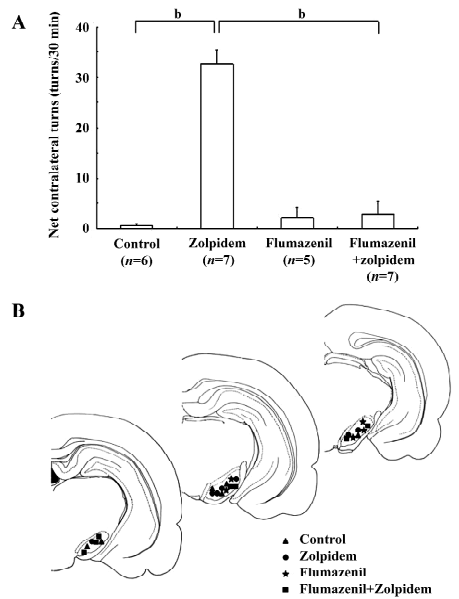
Discussion
It is well known that the subunit combination is critical in determining the physiological functions and pharmacological properties of the GABAA receptor in the brain[8,21–23]. Early morphological studies, including in situ hybridization, have shown that the SNr expressed high levels of α1 subunits on the GABAA receptor[7,24]. Zolpidem has high affinity for the GABAA receptor containing α1 subunits[25,26]. Therefore, neurons with relatively high densities of α1 subunits on the GABAA receptor usually exhibit high binding sites and sensitivity for zolpidem[27,28]. In the present study, all neurons recorded in the SNr exhibited sensitivity to 100 nmol/L zolpidem, suggesting the functional presence of α1 subunits in the rat SNr.
Morphological evidence has revealed that the SNr received GABAergic innervation from the striatum, globus pallidus, and local axon collaterals. The action potential-dependent inhibitory PSC of the SNr neurons mainly originate from the axon collaterals because the striatal neurons are quiescent. However, the action potential-dependent pallidal GABAergic inputs are blocked under coronal sectioning. To investigate the possible difference of the GABAA receptor subunit composition arising from striatonigral, pallidonigral, and nigronigral GABAergic innervation, we further compared the effects of zolpidem on mPSC and sPSC. The present results showed that zolpidem exerted similar potency on the prolongation of the decay time of mPSC and sPSC, suggesting that α1 subunit expression may be similar in the SNr. However, it was impossible to isolate the PSC arising from different sources and examine the effects of zolpidem on them directly.
Much evidence has shown that the SNr is involved in the control of several kinds of epileptic seizures. The firing rate of SNr increased significantly at the beginning of seizure[29]. The anticonvulsant drug gabapentin decreased the firing rate of the SNr[30]. Furthermore, deep brain stimulation of the SNr prevents seizures[31].
GABAergic transmission in the SNr is critical for seizure control. Previous studies have shown that GABAA receptor expression is reduced in the SNr of some epileptic models[32]. However, a recent study revealed that the GABAA receptor mediated inhibitory PSC, and α subunit expression were not reduced in the SNr of gerbils with inherited epilepsy[33]. Functional studies have shown that a microinjection of muscimol into the SNr suppressed the electroshock model of epilepsy[34], while bicuculline infusions are proconvulsant[35]. The intranigral transplantation of GABA-producing cells have been shown to exhibit significant anticonvulsant effects in experimental epilepsy[36]. Recently, Gonzalez Ramirez et al reported that benzodiazepine binding was enhanced in the SNr in hyperthermia-induced seizure rats[37]. Microinfusions of zolpidem had anticonvulsant effects on clonic and tonic-clonic seizures[13]. The present findings prompted us to hypothesize that the direct potentiation on GABA transmission is the major mechanism of zolpidem-induced antiepileptic effects in the SNr.
In conclusion, the present in vitro studies demonstrated that zolpidem enhanced GABA transmission in the SNr by activating the benzodiazepine site. Moreover, the finding on the effects of zolpidem in the SNr may provide a rationale for further investigations into its potential in the treatment of some neurological diseases.
References
- Ottersen OP, Storm-Mathisen J. Glutamate- and GABA-containing neurons in the mouse and rat brain, as demonstrated with a new immunocytochemical technique. J Comp Neurol 1984;229:374-92.
- Smith Y, Bolam JP. The output neurones and the dopaminergic neurones of the substantia nigra receive a GABA-containing input from the globus pallidus in the rat. J Comp Neurol 1990;296:47-64.
- Nitsch C, Riesenberg R. Immunocytochemical demonstration of GABAergic synaptic connections in rat substantia nigra after different lesions of the striatonigral projection. Brain Res 1988;461:127-42.
- Lantin le Boulch N, Truong-Ngoc NA, Gauchy C, Besson MJ. In vivo release of newly synthesized [3H]GABA in the substantia nigra of the rat: relative contribution of GABA striato-pallido-nigral afferents and nigral GABA neurons. Brain Res 1991;559:200-10.
- Somogyi P, Bolam JP, Smith AD. Monosynaptic cortical input and local axon collaterals of identified striatonigral neurons. A light and electron microscopic study using the Golgi-peroxidase transport-degeneration procedure. J Comp Neurol 1981;195:567-84.
- Nicholson LF, Faull RL, Waldvogel HJ, Dragunow M. The regional, cellular and subcellular localization of GABAA/benzodiazepine receptors in the substantia nigra of the rat. Neuroscience 1992;50:355-70.
- Wisden W, Laurie DJ, Monyer H, Seeburg PH. The distribution of 13 GABAA receptor subunit mRNAs in the rat brain. I. Telencephalon, diencephalon, mesencephalon. J Neurosci 1992;12:1040-62.
- Mehta AK, Ticku MK. An update on GABAA receptors. Brain Res Brain Res Rev 1999;29:196-217.
- Niddam R, Dubois A, Scatton B, Arbilla S, Langer SZ. Autoradiographic localization of [3H]zolpidem binding sites in the rat CNS: comparison with the distribution of [3H]flunitrazepam binding sites. J Neurochem 1987;49:890-9.
- Chadha A, Dawson LG, Jenner PG, Duty S. Effect of unilateral 6-hydroxydopamine lesions of the nigrostriatal pathway on GABA(A) receptor subunit gene expression in the rodent basal ganglia and thalamus. Neuroscience 2000;95:119-26.
- Pan HS, Penney JB, Young AB. Gamma-aminobutyric acid and benzodiazepine receptor changes induced by unilateral 6-hydroxydopamine lesions of the medial forebrain bundle. J Neurochem 1985;45:1396-404.
- Mereu G, Carcangiu G, Concas A, Passino N, Biggio G. Reduction of reticulata neuronal activity by zolpidem and alpidem, two imidazopyridines with high affinity for type I benzodiazepine receptors. Eur J Pharmacol 1990;179:339-45.
- Veliskova J, Loscher W, Moshe SL. Regional and age specific effects of zolpidem microinfusions in the substantia nigra on seizures. Epilepsy Res 1998;30:107-14.
- Zhang H, Rosenberg HC, Tietz EI. Anticonvulsant actions and interaction of GABA agonists and a benzodiazepine in pars reticulata of substantia nigra. Epilepsy Res 1991;8:11-20.
- Daniele A, Albanese A, Gainotti G, Gregori B, Bartolomeo P. Zolpidem in Parkinson’s disease. Lancet 1997;349:1222-3.
- Ruzicka E, Roth J, Jech R, Busek P. Subhypnotic doses of zolpidem oppose dopaminergic-induced dyskinesia in Parkinson’s disease. Mov Disord 2000;15:734-5.
- Farver DK, Khan MH. Zolpidem for antipsychotic-induced parkinsonism. Ann Pharmacother 2001;35:435-7.
- Chan PK, Yung WH. Inhibitory postsynaptic currents of rat substantia nigra pars reticulata neurons: role of GABA receptors and GABA uptake. Brain Res 1999;838:18-26.
- Yung WH, Hausser MA, Jack JJ. Electrophysiology of dopaminergic and non-dopaminergic neurones of the guinea-pig substantia nigra pars compacta in vitro. J Physiol 1991;436:643-67.
- Hausser MA, Yung WH. Inhibitory synaptic potentials in guinea-pig substantia nigra dopamine neurones in vitro. J Physiol 1994;479:401-22.
- Costa E. From GABAA receptor diversity emerges a unified vision of GABAergic inhibition. Annu Rev Pharmacol Toxicol 1998;38:321-50.
- Rudolph U, Crestani F, Mohler H. GABA(A) receptor subtypes: dissecting their pharmacological functions. Trends Pharmacol Sci 2001;22:188-94.
- Sieghart W. Structure and pharmacology of gamma-aminobutyric acidA receptor subtypes. Pharmacol Rev 1995;47:181-234.
- Duncan GE, Breese GR, Criswell HE, McCown TJ, Herbert JS, Devaud LL, et al. Distribution of [3H]zolpidem binding sites in relation to messenger RNA encoding the alpha 1, beta 2 and gamma 2 subunits of GABAA receptors in rat brain. Neuroscience 1995;64:1113-28.
- Macdonald RL, Olsen RW. GABAA receptor channels. Annu Rev Neurosci 1994;17:569-602.
- Perrais D, Ropert N. Effect of zolpidem on miniature IPSCs and occupancy of postsynaptic GABAA receptors in central synapses. J Neurosci 1999;19:578-88.
- Criswell HE, McCown TJ, Moy SS, Oxford GS, Mueller RA, Morrow AL, et al. Action of zolpidem on responses to GABA in relation to mRNAs for GABA(A) receptor alpha subunits within single cells: evidence for multiple functional GABA(A) isoreceptors on individual neurons. Neuropharmacology 1997;36:1641-52.
- Vicini S, Ferguson C, Prybylowski K, Kralic J, Morrow AL, Homanics GE. GABA(A) receptor alpha1 subunit deletion prevents developmental changes of inhibitory synaptic currents in cerebellar neurons. J Neurosci 2001;21:3009-16.
- Deransart C, Hellwig B, Heupel-Reuter M, Leger JF, Heck D, Lucking CH. Single-unit analysis of substantia nigra pars reticulata neurons in freely behaving rats with genetic absence epilepsy. Epilepsia 2003;44:1513-20.
- Bloms-Funke P, Loscher W. The anticonvulsant gabapentin decreases firing rates of substantia nigra pars reticulata neurons. Eur J Pharmacol 1996;316:211-8.
- Shi LH, Luo F, Woodward D, Chang JY. Deep brain stimulation of the substantia nigra pars reticulata exerts long lasting suppression of amygdala-kindled seizures. Brain Res 2006;1090:202-7.
- Olsen RW, Wamsley JK, McCabe RT, Lee RJ, Lomax P. Benzodiazepine/γ−aminobutyric acid receptor deficit in the midbrain of the seizure-susceptible gerbil. Proc Natl Acad Sci USA 1985;82:6701-5.
- Kumar SS, Wen X, Yang Y, Buckmaster PS. GABAA receptor-mediated IPSCs and alpha 1 subunit expression are not reduced in the substantia nigra pars reticulate of gerbils with inherited epilepsy. J Neurophysiol 2006;95:2446-55.
- Shehab S, Simkins M, Dean P, Redgrave P. Regional distribution of the anticonvulsant and behavioural effects of muscimol injected into the substantia nigra of rats. Eur J Neurosci 1996;8:749-57.
- Veliskova J, Velisek L, Nunes ML, Moshe SL. Developmental regulation of regional functionality of substantial nigra GABAA receptors involved in seizures. Eur J Pharmacol 1996;309:167-73.
- Loscher W, Ebert U, Lehmann H, Rosenthal C, Nikkhah G. Seizure suppression in kindling epilepsy by grafts of fetal GABAergic neurons in rat substantia nigra. J Neurosci Res 1998;51:196-209.
- Gonzalez Ramirez M, Orozco Suarez S, Salgado Ceballos H, Feria Velasco A, Rocha L. Hyperthermia-induced seizures modify the GABA(A) and benzodiazepine receptor binding in immature rat crain. Cell Mol Neurobiol 2007;27:211-27.