
The interaction of small molecules with phospholipid membranes studied by 1H NOESY NMR under magic-angle spinning1
Introduction
Pharmacologically active drugs are usually small molecules that target a specific protein, for instance an enzyme, a receptor, or a channel. Often, these molecules target membrane-stemming proteins, which represent about one third of all proteins in the human genome. It is estimated that on the order of 50%–70% of the current targets for drug development are proteins that are embedded in the cellular membrane[1,2]. The plasma membrane represents an important biological interface, where many essential processes for the cell function take place. Most importantly, it represents a barrier separating the cytosol from the extracellular medium. For specific functions, proteins are embedded in the membrane, where they are involved in signal generation and transduction, cellular communication, both active and passive transport of ions and a number of different molecules, and the generation of energy. In order to fulfill its function, a drug molecule has to find its specific protein interaction partner and specifically bind to it. Although about 60% of the dry weight of the membrane is constituted of proteins, due to the large variety of these molecules the actual concentration of a specific protein in the membrane is typically relatively low, which decreases the likelihood for a drug to find its target protein in the membrane. However, the binding of a drug to its target can be facilitated by initial partitioning into the membrane and subsequent binding to its target protein[3]. Such a scenario can have several advantages. First, pharmacological drugs are often lipophilic molecules that preferentially partition into a lipid membrane or its interface. Second, the diffusion of a drug in the membrane is constraint to two dimensions, which increases the interaction probability between membrane protein and its ligand by approximately a factor of 1000[4]. Third, the area of the membrane is much larger than that of the specific protein. Therefore, unspecific binding of a drug molecule to the membrane will bring it in close proximity to its target. Since the binding constant of the drug to the target protein is usually larger than to the membrane, subsequent protein binding will necessarily occur. These arguments may suggest a mechanism of membrane partitioning of a drug molecule prior to binding to its target protein.
To date, only a few mammalian membrane proteins are known in their structure to high resolution[5,6]. As for membrane proteins, the current understanding of the interaction of small molecules with the plasma membrane is also limited. Because of the complicated membrane structure, membrane proteins are difficult to crystallize in their natural environment. A biological membrane is characterized by a versatile molecular dynamics that takes place in a broad time window from picoseconds to hours[7-10]. Further, biological membranes feature a highly specialized lipid-water interface that allows for numerous intermolecular interactions to take place to stabilize a protein in its correct structure[11-13]. Likewise, the interactions of small lipophilic molecules such as drugs with a membrane are diverse and complicated usually giving rise to a broad and dynamic distribution of them. A sketch of the complicated membrane structure and the distribution of molecules within the lipid-water interface is provided in Figure 1. Considering the complicated nature of a biological membrane and its lipid-water interface, simple partition experiment in water/octanol biphasic systems do not provide good models for drug partitioning into membranes and more structure-based biophysical methods are required to understand binding of small molecules to the membrane interface.
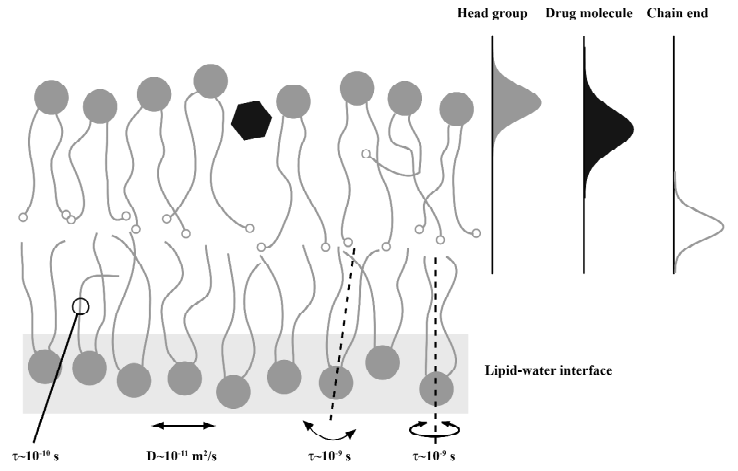
In lipid membranes, all molecules are affected by very complex interaction patterns brought about by basic electrostatic, hydrophobic, and van der Waals interactions arising from the physical and chemical features of the interacting molecules. Also, hydrogen bond formation, lipid induced interactions, and a multitude of entropic contributions play an important role for the interaction between the constituents in the bilayer. At any given depth of a membrane, different interaction patterns will dominate leading to very specific physical properties on a scale of only several Angstroms along the membrane normal. A small molecule that partitions into the bilayer is subject to large gradients of the basic physical parameters such as dielectric constant, water content and so on giving rise to very specific and site-resolved interactions. The average location of a molecule in the membrane is consequently determined by a minimum in the Gibbs free energy that contains both entropic and enthalpic contributions. But it needs to be stressed, that in most cases membrane embedded molecules are broadly distributed around their average location and subject to high dynamics on different time scales[14,15]. In such a complex environment a high spatial resolution for the membrane position and orientation of a specific molecular segment is required to understand the interactions between different molecules and to relate this to the molecular function. Combined neutron and x-ray scattering techniques may achieve such a resolution, which requires isotopic labeling and the preparation of oriented samples. Typical, scattering experiments perform better with dehydrated samples, which could be intrusive[15,16]. Over the last 20 years, an 1H NMR technique became available that allows to measure intermolecular contact probabilities in lipid membranes allowing to determine the distribution profiles of small molecules[17].
This review will focus on 1H solid-state NMR applications to study the interaction of small molecules with membranes and to determine their distribution within the bilayer. Solid-state NMR can not only provide important contributions for solving the structure of membrane proteins[8,18-20] but also represents a very useful technique to study the lipid membrane itself[10,21-24]. In particular, the interaction of small molecules such as natural and synthetic drugs with membranes can be very successfully studied by solid-state NMR techniques[25-27]. Studies about membrane binding, the response of the lipid membrane to the presence of such molecules, the interactions between different molecules, the lateral membrane organization, and the membrane diffusion are possible. Such information is crucial to understand the function and the complicated interplay of membrane molecules and the entire membrane and may help to identify target structures for drug research or to provide insight into the mode of action of the investigated molecules.
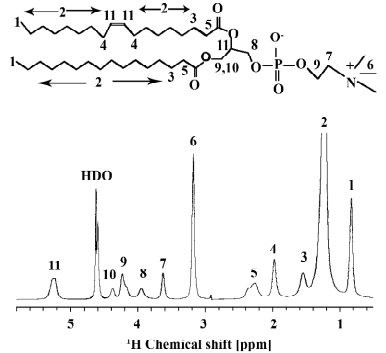
Usually, solid-state NMR spectra are characterized by broad anisotropic line shapes. However, the magic-angle spinning (MAS) technique can convert broad anisotropic signals into well-resolved sharp lines[28]. For this technique, the NMR sample is contained in a cylindrical MAS rotor that is oriented at an angle of 54.73º with respect to the external magnetic field and spun about its long axis at an angular frequency of up to tens of kHz. Usually, for well-resolved 1H NMR spectra the MAS frequency has to exceed the width of the 1H-1H dipolar tensor interaction. However, for lipid molecules in the fluid phase of the membrane, well-resolved 1H MAS spectra could be recorded at low MAS frequencies although the homogeneous broadenings are on the order of 10 kHz and larger[17]. This effect has been explained by the rapid axially symmetric reorientation of the lipids in the membrane with a correlation time on the order of 1 ns or less[29]. Thus, the dipolar broadening only depends on the orientation of the local bilayer normal relative to the external magnetic field scaling with the second Legendré polynomial P2(cosθ). In this manner, the homogenous dipolar broadening is converted into an inhomogeneous broadening, which can be averaged out by MAS even if the rotational frequency is lower than the width of the anisotropic spectrum[29]. Typical line widths of 1H MAS NMR spectra of lipid membranes are on the order of 0.01–0.1 ppm. An example of a 1D 1H MAS NMR spectrum of an aqueous suspension of POPC with the assignments of all lipid signals is shown in Figure 2.
The good resolution of 1H MAS NMR spectra of lipid membranes and their long spin-lattice relaxation times allows acquisition of two dimensional correlation spectra for the study of intermolecular interactions. This review is devoted to 1H MAS nuclear Overhauser enhancement spectroscopy (NOESY) NMR investigations of lipid membranes and their interaction with small molecules -from water to peptides- incorporated in lipid membranes. While the NOESY technique in solution NMR is mostly applied to determine intramolecular distances in soluble protein or DNA molecules, for the application to lipid bilayers the intermolecular interactions are most interesting. Determination of intermolecular cross-relaxation rates allows to study membrane binding, membrane organization, membrane localization and membrane orientation of membrane embedded molecules. In several studies 1H MAS NOESY NMR has shown its high potential in such investigations.
Theory
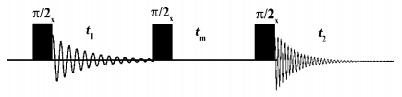
Interpretation of intermolecular cross-relaxation rates in membranes The simple and robust three pulse NOESY pulse sequence is shown in Figure 3[30,31]. The first pulse creates transversal magnetization, which is allowed to evolve under the influence of the isotropic chemical shift during t1. Thus, each spin is labelled with its characteristic resonance frequency. The second pulse flips the magnetization into the z-direction, where magnetization exchange takes place during the mixing time tm. This leads to the build up of cross peaks in the 2D spectra provided the interacting spins are in close proximity during mixing (< 5 Å). Since chemical exchange typically does not contribute to the magnetization exchange in lipid membranes the cross peaks are solely caused by dipolar cross-relaxation between two proton spins in close proximity. Such a process can be described by the cross-relaxation rate σ, which is strongly dependent on the distance r between the two spins and the correlation time of the molecular motion τc according to[32,33]:
In equation (1), η is Planck’s constant divided by 2π, μ0 is the magnetic constant, ω0 is the Larmor frequency, and γ the gyromagnetic ratio. Due to this very strong distance dependence only interactions contribute to the cross-relaxation rate that occur within 5 Å. If NOESY experiments are used to study the interaction of small molecules with lipid membranes, only intermolecular cross-relaxation is of interest. This is different from the situation encountered for soluble proteins, where cross-relaxation rates provide a direct measure of the inter-proton distance provided the correlation time that modulates the interaction is known. In contrast, intermolecular cross-relaxation rates determined in lipid membranes in the fluid phase state, can not be used to measure a well defined fixed distance between the regarding protons. Due to the high mobility and molecular disorder in lipid membranes[14] the cross-relaxation rates provide a measure of the contact probability between the interacting spins[34,35]. Therefore, in a lipid membrane the cross-relaxation rate is proportional to the contact frequency between the two nuclei, a high contact frequency results in large cross-relaxation rates and a low contact frequency yields small cross-relaxation rates.
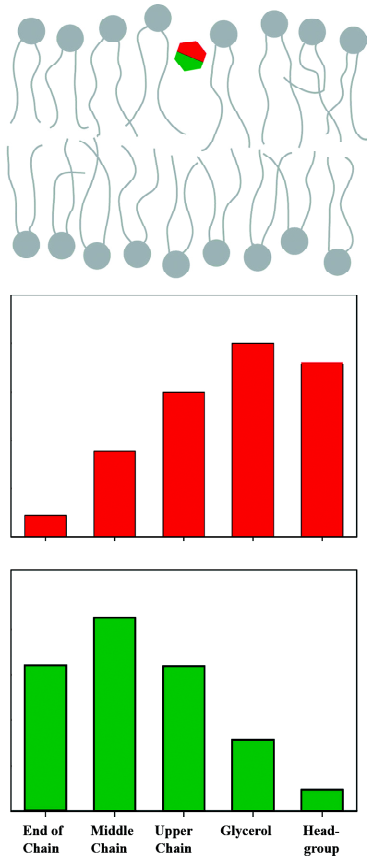
The contact probability, ie, the cross-relaxation rate between two nuclei, is directly related to the structural arrangement in the membrane. If we assume a broad distribution function for each molecular segment, it becomes obvious that for some of those segments the distribution functions overlap and for others they do not. A strong overlap in the distribution functions leads to large cross-relaxation rates, small overlap creates small cross-relaxation rates, and if there is no overlap in these distribution functions, no cross-relaxation rate is measured. Thus, cross-relaxation rates in membranes represent a measure for the interaction strength between molecular segments, which can be used to localize molecules in the membrane and to study the lateral molecular interaction. For small membrane embedded molecules cross peaks with more than just one lipid segment are observed, often even all lipid segments exchange magnetization with the molecule of interest. This is related to the high degree of molecular disorder and the high mobility of the molecules in the membrane[35]. Therefore, a plot of the cross-relaxation rates of a particular molecular segment vs. all lipid segments along the membrane normal describes the distribution function of this segment in the lipid bilayer as sketched in Figure 4. Further, if the distribution functions of two or more spins of the membrane embedded molecule are known, one can typically obtain the membrane orientation of this compound.
It should be mentioned that this interpretation of NOESY cross-relaxation rates is only correct if the correlation time that modulates the magnetization exchange is equal for all the different molecular contacts. First estimations of the correlation times that are responsible for the magnetization exchange in a NOESY experiment in membranes revealed that only relatively slow processes have to be considered. These slow processes describe motions of entire molecules[7] and lateral lipid diffusion[36], which suggests that all molecular contacts can be treated equally and the NOESY cross-relaxation rate indeed represents the contact probability between molecular segments.
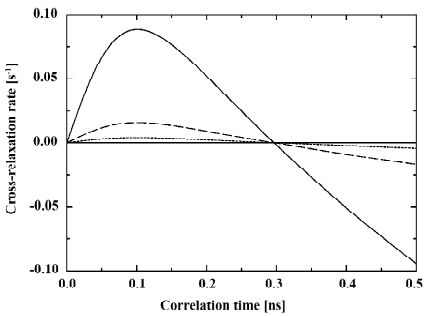
To avoid over-interpretation of the NOESY data care should be taken in comparing absolute cross-relaxation rates for different molecules in lipid membranes. The dependence of the cross-relaxation rate σ on the correlation time of motion τc in equation (1) may lead to remarkable changes in the cross-relaxation rates even for small variations of the correlation time as shown in Figure 5. Even though all cross-relaxation is approximately modulated by the same correlation time, very small alterations can lead to significant changes in the cross-relaxation rates.
Determination of cross-relaxation rates in membranes There are three possibilities to calculate cross-relaxation rates from 2D NOESY spectra: the full matrix approach, the spin pair model, and the single mixing time approach[35].
The most complex and time consuming but also the most accurate method is the full matrix approach. The rate of magnetization transfer dΔIZk/dtm of the proton k either to the lattice (spin-lattice relaxation rate ρkk) or to other protons j of the spin system (cross-relaxation rate σkj) is described by the extended Solomon equation[32]:
This can be written down for each spin in the system leading to a set of differential equations. A measure for the transferred magnetization is the volume of all diagonal and all cross peaks of a 2D NMR NOESY spectrum at a given mixing time tm arranged in the matrix A(tm):
The peak volumes in 2D NOESY spectra are typically obtained from numerical integration routines. The relaxation matrix R contains all spin-lattice and cross-relaxation rates for a system of N spins:
Most accurately, the relaxation matrix is calculated by fitting equation (3) to the experimental peak volume matrices obtained for different mixing times. The resulting relaxation matrix contains all parameters to determine the localization and the orientation of a given molecule in the lipid membrane.
Alternatively, the spin-pair interaction model assumes that the spin system is reasonably well decoupled and reduces the multi-spin system to an isolated spin pair. The volume of a cross peak between the two spin I and S is then obtained by[37]:
where AIS(tm) represents the cross peak volume at the mixing time tm, AII(0) the volume of the diagonal peak at tm=0, and T1,IS defines the magnetization leakage to the lattice. By analyzing the cross peak volumes for different mixing times (and the diagonal peak volumes for a very short mixing time) the cross-relaxation rate can be obtained from a simple fit to this equation. The advantage of the spin-pair-model is that only the cross peaks between the investigated molecule and the molecular segments of the lipid matrix need to be integrated to calculate the respective cross-relaxation rates. Since the determination of the complete peak volume matrices is tedious and time consuming, this approach is more efficient if only a few cross-relaxation rates are required for a certain application. Though the information of the complete relaxation matrix is lost, it is not necessary to monitor all magnetization transfer processes within the lipid matrix, if one is only interested in the localization of a molecule in the membrane.
The most simplistic single mixing time approach determines cross-relaxation rates from a single NOESY experiment conducted at just one mixing time. In this approach, equation (5) is expanded into a truncated Taylor series that only contains the linear term, which leads to[35]:
This simplification represents the most efficient approach since only a single NOESY spectrum needs to be acquired to determine cross-relaxation rates. However, the cross-relaxation rates determined by this approach may not be very accurate, in particular small rates can be highly biased when there is also strong (intramolecular) magnetization exchange of the particular segment with other molecular sites.
The three approaches for the determination the cross-relaxation rates from 2D NOESY spectra were compared in an DMPC lipid matrix[35]. For the majority of the cross-relaxation rates, reasonable agreement between the three models was reported. However, the cross-relaxation rates between neighboring protons in lipid hydrocarbon chains are very high, which interferes with the calculation of magnetization transfer between chain protons, which are more distant. This situation is only correctly reflected in the full matrix approach.
1H NOESY MAS NMR of pure lipid membranes
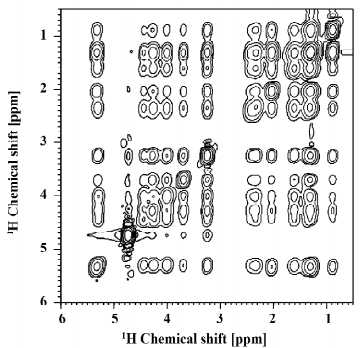
Before discussing 1H MAS NOESY NMR investigations of the interaction of small molecules with lipid membranes, we will first briefly review NOESY data on pure phospholipid bilayers that provided insight into their highly dynamic organization and molecular disorder. The most extensive studies were done by the Gawrisch group, who investigated the lateral lipid organization in pure DMPC bilayers[35] and complex phospholipid mixtures in the presence of cholesterol[38]. A typical 1H MAS NOESY spectrum of lipid bilayers is shown in Figure 6. Interestingly, cross peaks between all lipid segments of the phospholipid molecules were observed due to the high mobility and molecular disorder in lipid membranes. This could only be understood from a quantitative magnetization transfer analysis within the lipid membrane using the full matrix approach[35]. Experiments in mixed perdeuterated/protiated phospholipid membranes indicated that the cross-relaxation in lipid membranes is almost entirely of intermolecular origin. Therefore, the occurrence of NOESY cross peaks between all lipid segments can only be explained by a membrane model that is characterized by a high degree of motional disorder. Indeed, if per proton based cross-relaxation rates were compared, the most likely contacts between molecular segments also showed the highest cross-relaxation rates. Less frequent contacts, for instance those between lipid headgroups and the ends of the acyl chains produced smaller cross-relaxation rates. Although such contacts are by far less frequent, they occur with a relative high likelihood in a liquid-crystalline membrane indicating the high amount of disorder and mobility in biological membranes.
1H NOESY cross-relaxation rates were also determined in mixed membranes of polyunsaturated SDPC/SDPE/SDPS/Cholesterol (4/4/1/1, mol/mol) membranes[38]. Since cross-relaxation rates can be determined for intermolecular lipid-lipid contacts, they are ideal tools to study lateral lipid organization. In particular, the interaction of cholesterol with the unsaturated lipid species was studied. The most important results of this study were that (i) cholesterol preferentially interacts with the SDPC in the mixture, while lateral contacts between cholesterol and SDPE and SDPS are less frequent and (ii) cholesterol prefers contacts with saturated chains, where favorable van der Waals interactions between the chains and the rigid sterol ring systems are formed. In contrast, interactions of cholesterol and the polyunsaturated chains were less frequently observed. These results allowed to determine a quantitative model of the lateral lipid organization in this complex mixture, which revealed the existence of microdomains that are enriched in SDPC and cholesterol[38].
So far, the issue of spin diffusion has not been discussed. Spin diffusion represents an undesired mechanism that relays magnetization over several steps to yield a cross peak for molecular segments that are not in close proximity. Since this relayed transfer of magnetization is typically slow, spin diffusion can be neglected if the mixing time is kept to a minimum. Since the occurrence of head-to-tail cross peaks in NOESY spectra of lipid membranes was previously attributed to spin diffusion[39,40], this mechanism was investigated in more detail[41]. In particular, special phospholipid molecules were employed that contained two deuterated methylene groups in the middle of the chain to block spin diffusion. The quantitative analysis of cross-relaxation rates in this study proved that the cross peaks between lipid headgroups and hydrocarbon chains are not caused by spin diffusion but are the result of a direct approach (within 5 Å) of headgroup and chain protons[41]. Again, only highly disordered and dynamic membranes can explain this result.
NOESY MAS NMR on lipid membranes containing small molecules
Several aspects of the interaction of small molecules with lipid membranes were investigated by 1H MAS NOESY NMR studies over the last years. Starting with the question of how deep water penetrates a lipid membrane; further studies investigated the membrane interaction of ethanol and different aromatic molecules. Finally, 1H MAS NOESY NMR was used to study the membrane location and interaction of peptides and lipophilic nucleosides. In addition, several fluorescent probes like lipid and cholesterol analoga were investigated in a membrane environment.
Water The water distribution in a lipid bilayer is an interesting problem that was one of the first issues investigated by 1H NOESY MAS NMR. Specific interactions between water and the lipids are essential for the formation of different lipid phases and in particular planar lipid membranes[27]. At the same time, the water permeation across bilayers is very high[42]. Therefore, the main questions concerned the depth water penetration into the bilayer and the thickness of the lipid-water interface of the membrane. A number of studies measured intermolecular cross-peaks between water and the individual lipid segments[43-46].
Phase-sensitive NOESY experiments were applied under MAS conditions to show that the water molecules have access to the headgroup, the glycerol backbone and the upper chain region of a POPC bilayer[43]. The loss of some of these cross peaks in the presence of a non-ionic detergent (C12EO4) indicated a process of dehydration due to the presence of the surfactant.
Further NOESY studies on lipid hydration of MeDOPE and DOPC bilayers[35] showed that the expected cross peaks between water and lipid headgroup/glycerol are detected for MeDOPE but are absent for DOPC membranes and even in mixed MeDOPE/DOPC bilayers[45]. Also for the hexagonal and the metastable isotropic phase of MeDOPE the lipid-water cross peaks were not observable, indicating that in this phases the water is diffusing freely and no longer involved in the hydrogen bonding pattern at the lipid headgroups[43]. However, the presence or absence of cross-peaks between lipids and water should be interpreted with great caution. Since the lifetime of water-lipid associations is likely to be shorter than of lipid-lipid associations, those cross-peaks are often weak and have negative intensity. They are easily missed if spectra are not recorded at proper experimental conditions. This is underlined by the fact that in the same study heteronuclear HOESY experiments indicated contacts between the DOPC headgroups with the phosphate buffer (used as reporter for the water location)[45]. Therefore, water-lipid NOESY cross peaks could have been below the detection limit due to their weak intensities. On the other hand, molecules that exchange protons with water via hydroxyl and amino groups may show strong cross-peaks to water. However, those peaks may reflect chemical exchange of protons.
In the most recent study the cross-relaxation rates between water and phospholipid segments were determined in a full matrix approach for POPC[46]. While cross-relaxation rates in lipid membranes are always negative, lipid-water cross-relaxation rates were calculated to be positive indicating faster correlation times for the interaction on the order of 100 ps. This value agrees well with the short lifetime of water in the first hydration layer[47]. Water showed weak cross-relaxation rates with the signals from the lipid headgroup and the glycerol. Only very low cross-relaxation rates were determined for the acyl chain region of the membrane suggesting that the water concentration in the bilayer core is very low.
Taken together, these results indicate that there are a number of water molecules in the lipid-water interface that are in close proximity to the lipid headgroups, the glycerol region, and the carbonyl groups of the acyl chains. Here, favorable hydrogen bonds are formed that give rise to the observed NOESY cross peaks. All studies exhibited that there is no water detectable in the hydrophobic core of membrane. The water permeation across bilayers is very high[42]. However, the individual act of a water molecule passing through the bilayer is likely to require only a fraction of a microsecond. Therefore, water content in the hydrophobic core of a bilayer at any given time is rather low, which is in agreement with observations by neutron scattering.
Ethanol The interaction of ethanol with lipid membranes has been a controversy in anesthesia for several years. Some studies report that the effect of ethanol on membrane proteins is attributed to changes in the lipid packing of the membrane[48], while others emphasize it is the result of ethanol binding to the membrane protein itself [49]. While the molecules could interact at multiple sites, 1H MAS NMR has been used to determine the exact localization and distribution of ethanol in the lipid membrane by Gawrisch and co-workers[50,51]. In these studies, relative cross peak intensities and later cross-relaxation rates were for the first time interpreted in terms of a distribution function of the molecule relative to various molecular segments of the lipids and over the lipid bilayer[50]. In these profiles, ethanol showed contacts to all phospholipid segments due to the high disorder and mobility in liquid-crystalline membranes. Being a small amphiphilic molecule, ethanol was localized in the lipid-water interface of the membrane, with the largest cross-relaxation rates (ie, contact probabilities) to the glycerol backbone. The degree of chain unsaturation in the lipid matrix only had a slight effect on the membrane distribution of ethanol as shown for saturated DMPC, monounsaturated SOPC, and polyunsaturated SDPC bilayers[50]. A further study of the ethanol distribution in POPC membranes revealed an identical position of ethanol in the bilayer and showed excellent agreement between the measured cross-relaxation rate profile and this profile calculated from a molecular dynamics simulation[51]. In addition, a comparison of the ethanol distribution profiles exhibited that the methyl group of ethanol is more deeply inserted into the membrane then its methylene group, revealing that the hydroxyl group as the most polar part of the molecule is pointing towards the membrane surface.
In conclusion, ethanol is localized in the chemically and physically very heterogeneous region of the lipid-water interface of the membrane interacting with the lipid molecules mainly by hydrogen bonds and smaller hydrophobic contributions. It is expected that ethanol interacts via similar mechanisms with protein surfaces and influences protein function.
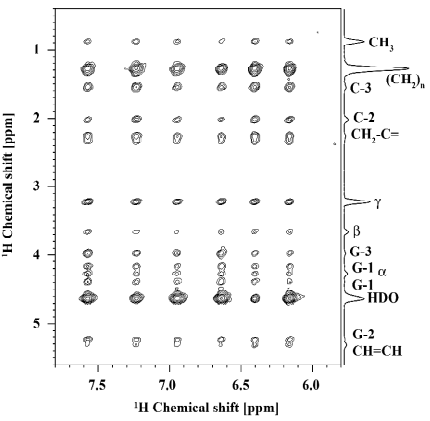
Small aromatic compounds One big challenge in the analysis of 2D NOESY spectra for the localization of small molecules in the lipid membrane is to unambiguously distinguish between cross peaks that are caused by the interactions of lipids with the small molecules and the less interesting lipid-lipid cross peaks. Usually, the latter are much higher in intensity rendering a quantitative interpretation of the cross-relaxation rates challenging. The above mentioned lipid-ethanol mixtures are an example for these problems, because lipid-ethanol cross peaks are superimposed with the lipid-lipid signals[50]. For this reason, aromatic compounds interacting with lipid membranes are much easier to study. The chemical shift dispersion of the aromatic resonances lies outside that of the phospholipids (> 6 ppm) leading to NOESY NMR spectra that can be easily analyzed. A very clear example is given in Figure 7 for a luteolin/POPC preparation. Here, the risk of misreading cross-peak volumes because of signal superposition with lipids is abolished. This renders the method very well suited for the determination of pharmaceutical drugs, as these compounds are often aromatic.
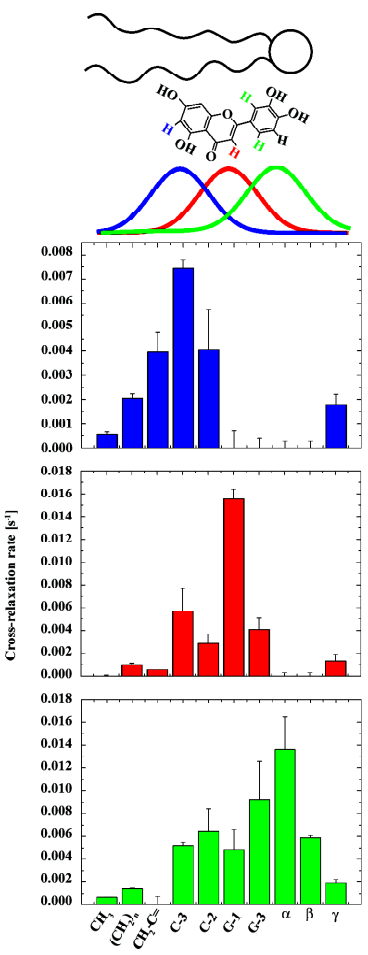
The membrane interaction of a selection of flavonoids (flavone, chrysin, luteolin, myricetin) were investigated by Scheidt et al[52]. Flavonoids are small aromatic compounds considered to be beneficial for human health. A long list of biological activities is ascribed to these molecules, such as an antioxidative effect, which is depending on their interactions and the penetration depth in lipid membranes. By extensive 1H MAS NOESY NMR studies, the distribution profiles of the flavonoid molecules in the membrane were determined. Again these molecules exhibited relative broad distribution profiles in the membrane, which was explained by the high mobility and molecular disorder. Subtle differences in the membrane localization of the flavonoids studied were revealed by the technique. These differences originated from polarity alterations in the molecules due to a varying number of hydroxyl groups. In the complex interaction pattern of lipid membranes even the small changes in the chemical structure between the molecules led to changes in the physical interactions and influence the membrane position. Also, the orientation of a molecule within the membrane could be examined by comparison of the cross-relaxation rate profiles of different protons on one flavonoid molecule. These led to distribution functions of the flavonoid relative to the lipid bilayer as shown for luteolin in a POPC membrane in Figure 8. These orientations of the flavonoid molecules in the complex chemical and physical environment of a lipid membrane are also determined by the localization of the OH-groups in the molecule -the most polar part of the molecule is exposed to the aqueous phase. In addition, such changes were shown for luteolin-7-glucoside. Flavonoid glucosylation is very common in nature, this process modifies the molecular properties (polarity, size) quite significantly and lead in this case to a reverse membrane insertion of luteolin-7-glucoside compared to luteolin. These results indicated that flavonoid molecules could access all lipid segments and have the ability to contact different parts of the membrane (especially the double bonds of the lipid chains) to exert their antioxidative effects.
Aromatic molecules with a rather similar chemical structure were investigated by 1H MAS NOESY NMR in the Glaubitz group[53]. Here, nine substrates and modulators of the ABC transporter P-Glycoprotein (PGP) (including penicillin G and the flavonoid quercetin) were studied systematically in DMPC membranes to gain insight into their membrane location and interaction with lipid molecules. These molecules belong to distinct classes such as Ca2+ channel blockers, cytotoxic agents, antihypertensives, anthracyclines, or bacterisostatics. Due to their lipophilic nature these drugs are accumulated in the membrane. By analyzing the NOESY cross peaks between signals of the aromatic finger print region of the substrates and the lipid resonances a membrane distribution in the glycerol and the upper chain region was found for all investigated molecules. Again, the small differences in the distribution profiles were caused by alterations in the polarity between these molecules. The location of all structurally different molecules in the membrane interface indicated that the PGP binding site, which is assumed to be in the transmembrane region of PGP, is likely accessible. Drug binding could take place by cation-π[54] and π-π-stacking interactions with the aromatic and polar residues of PGP. The high similarity between the distribution profiles led to the conclusion that the molecules use similar pathways of interaction with PGP.
A number of aromatic indole and indole analogues were studied by 1H MAS NOESY NMR to determine their membrane distribution [55]. The study was motivated by the fact that aromatic amino acids such as tyrosine and tryptophane are enriched in those parts of membrane proteins that are located in the lipid-water interface of the membrane. In order to study the preference of these compounds for the membrane interface four indole and indene molecules were studied as analogues for trytophane. For all compounds, the maximum of the cross-relaxation rate profiles and therefore the membrane distribution was found in the glycerol region. These results were supported by induced chemical shift data and neutron diffraction results[55] and could be confirmed for indole in different phospholipid matrixes [56]. The comparison of the molecules, which were systematically varied in their dipole moment and their ability to form hydrogen bonds, showed that the cation-π-interaction plays an important role in the interfacial interactions of such molecules. In the complex environment of a lipid membrane the balance of electrostatic, hydrogen bonding and cation-π-interaction determine the location of Trp in the interface influencing the membrane properties of peptides and proteins containing aromatic residues.
Peptides About one third of the human genome encodes membrane proteins, which are the major targets in current pharmaceutical research. Since structural data for mammalian membrane proteins is scarce, any information about the membrane interaction and location is of great interest. In particular for small segments of larger membrane proteins, 1H MAS NOESY NMR experiments can provide insights into structural and dynamical aspects of these molecules. Again, the strength of this experimental approach lies in the determination of the distribution profile of certain peptide segments with respect to the lipid membrane[57-61].
The model for the pore structure of gramicidin A in membrane environment was confirmed by observing NOESY cross peaks between the trytophane residues of gramicidin A, which are located at the C-terminal end of the peptide, and the lipid headgroups, the glycerol region and the upper chain of DLPC membranes. Additionally the formyl moiety exhibited the expected cross peaks with the methyl and methylene groups of the acyl chains due to its location in the center of the bilayer[57]. This study proved that the spatial selectivity of 1H MAS NOESY experiments is sufficient to gain information about membrane-peptide interactions.
Unstructured clusters of aromatic and basic residues are widely found in membrane proteins involved in signal transduction. The membrane interaction of such proteins is often mediated by these polybasic clusters. As an example for such proteins, a phenylalanine rich effector domain of the MARCKS protein (amino acids 151–175) was investigated in membrane environment using 1H MAS NOESY experiments by Zhang and co-workers[58]. The strong cross peaks between the aromatic resonances and the acyl chain protons of the lipids showed that the five Phe rings of MARCKS(151–175) are penetrating the membrane and are located below the acyl chain carbonyl groups. In this configuration, the positively charged amino acids of MARCKS(151–175) came also in close proximity to the negatively charged lipid phosphates, implying an important influence of electrostatic interactions on the membrane interaction of this peptide. These results could confirm previous EPR data that showed a membrane surface localization of MARCKS(151–175) with the Phe residues penetrating the membrane interface using saturation transfer EPR techniques[62].
Epand and co-workers used the relative strength of NOE cross peaks between aromatic residues and the phospholipids to study the modulation of the peptide-membrane interactions in the absence and in the presence of cholesterol[59-61]. This is of outstanding interest with regard to the raft hypothesis[63] and the influence of cholesterol on protein-lipid-interaction[64] and protein function[65,66]. In all studies, an influence of the cholesterol content in the lipid membrane on the 1H MAS NOESY cross peaks was shown. For the peptide LWYIK (from the HIV-1 fusion protein gp41)[59] and a peptide corresponding to the N-terminus of the neuronal protein NAP-22[60] a stronger peptide-membrane interaction and a deeper insertion of the aromatic residues were determined from of stronger cross peak intensities in the presence of cholesterol.
The 1H MAS NOESY technique was also used to determine a model for the membrane insertion of the C-terminus of the lipidated human N-Ras protein[67,68]. Ras is involved in one of the major signal transduction cascades in biology stimulating cell proliferation and differentiation. Only in the membrane bound state the Ras protein is active. In a series of papers, a doubly lipid modified heptapeptide constituting the membrane anchor of Ras was studied[67,68]. The observation of cross peaks between peptide and lipid signals not only proved the membrane binding of the Ras peptide - a careful analysis of the cross-relaxation rate profiles of different peptide signals led to a structural model for the insertion of the Ras peptide into a lipid membrane. According to this model, the peptide backbone is located in the lipid-water interface of the membrane, where it strongly interacts with the lipid molecules by dipole-dipole interactions and hydrogen bond formation. The hydrophobic peptide side chains and the two lipid modified Cys residues insert deeply into the apolar membrane interior. This model could be confirmed by FTIR and neutron diffraction measurements[68] and helped building a structural model of the membrane anchor of the entire Ras protein[69].
Fluorescent lipid and cholesterol analogs Another interesting application of 1H NOESY MAS NMR measurements is the study of the membrane interaction and localization of fluorescence labels and fluorescent lipid analogues. These molecules are used in several biophysical and biochemical studies to investigate intracellular trafficking or membrane organization. Such phospholipid or sterol molecules either carry a fluorescent probe that is covalently attached (mostly NBD) or in the case of sterol molecules become fluorescent due to changes in the molecular ring structure. For the application of such molecules it is very important to know exactly how well they mimic the behavior of their unmodified counterparts to interpret the data achieved using these analogues correctly. As seen above, 1H MAS NOESY NMR can deliver very useful information about the membrane localization and orientation of such molecules.
By analyzing the cross-relaxation rates between NBD groups covalently attached to the acyl chain end of phospholipid analoga and different molecular segments of the phospholipid matrix the distribution profile of the NBD group in the lipid bilayer was determined[70,71]. These profiles proofed that the NBD groups are located in the lipid-water interface and not in the hydrophobic core of the membrane as may be expected for a NBD group attached to the acyl chain end of a phospholipid molecule. The backfolding of the NBD moiety attached to the sn-2 chain of a PC molecule is illustrated in Figure 9. Various interactions between the charged bulky NBD group and the phospholipids (Coulombic, dipole-dipole, cation-π, and hydrogen bonds) lead to a backfolding of the acyl chain and place the NBD group in the membrane interface region, where it experiences a more favorable free energy. Therefore, the data achieved with such NBD labeled phospholipids has to be interpreted carefully. Further, due to the broad distribution of the NBD label in the membrane interface, the probe is not well suited for distance measurements with sub-Angstrom resolution. The same applies for the distribution of EPR probes[72].

In analogy to this study, the membrane orientation of cholesterol and its naturally fluorescent analogues dehydroergosterol and cholestatrienol were investigated[73]. For these molecules cross peaks between a methyl group at the ring structure and the different segments of the phospholipid molecules were observed. Very similar cross-relaxation rate profiles across the lipid bilayer were measured for all three sterols, where the hydroxyl group, as the most polar part of the molecule, is localized to the lipid-water interface of the membrane. Therefore, it was concluded that the membrane localization and orientation of cholesterol, dehydroergosterol and cholestatrienol is comparable.
Lipophilic nucleotides and nucleosides Lipophilic nucleotides and nucleosides are designed for the potential use in nanobiotechnology by combining the molecular recognition properties of nuclear acids with the self-assembly characteristics of lipids in planer surfaces. By linking nucleosides with a hydrophobic molecule, lipophilic molecules are created that can be used to functionalize membrane surfaces that display certain recognition patterns. By using complementary single stranded DNA molecules, the membrane can be functionalized to bind enzymes, drugs, or other molecules of interest that are covalently attached to the DNA. For this purpose, the lipophilic nucleotides have to be incorporated into the lipid membrane, they should be stably anchored, and the nuclear acid moiety should be easily accessible from the aqueous phase.
To confirm these properties and to optimize the design of the molecules 1H MAS NOESY NMR was used[74-77]. By observing intermolecular cross peaks between resonances of the lipophilic nucleotide and the phospholipid the membrane incorporation of such designed molecules was demonstrated. Further, the analysis of the cross-relaxation rates between different segments of the lipophilic nucleotide[77] or nucleoside[74,75] and the phospholipid segments revealed the distribution profiles of the molecules in the lipid membrane. For the lipophilic nucleosides, the nucleobases were found to be localized in the lipid-water interface of the membrane, which represents an unfavorable location of these groups because their accessibility for base pairing with complementary single stranded DNA would be limited[74].
Concluding remarks
The examples discussed in this article show that 1H MAS NOESY NMR is a useful method for the investigation of the lipid membrane structure, lateral lipid organization, and the localization of small molecules and molecular segments in lipid bilayers with atomic resolution. Taken together, the most surprising results of 1H MAS NOESY studies are the broad distributions that are determined for small molecules localized in lipid membranes. However, these effects are confirmed by recent x-ray studies[14,15,78] and molecular dynamics simulations[9,79] that picture lipid membranes as highly dynamic and thermally disordered two-dimensional liquids. Consequently, molecules that bind to the membrane and all segments of the molecules constituting a biological membrane are subject to broad distribution functions and are by no means constraint to a single fixed position. This highly dynamic view of molecular processes that take place in the lipid environment should be adapted to the models that are used to explain complex processes such as lipid-protein interaction, membrane protein structure, and the interaction of membrane proteins with their ligands and drug molecules.
Abbreviations
DMPC 1,2-dimyristoyl-sn-glycero-3-phosphocholine
DLPC 1,2-dilauroyl-sn-glycero-3-phosphocholine
DOPC 1,2-di-oleoyl-sn-glycero-3-phosphocholine
HOESY heteronuclear Overhauser enhancement spectros copy
MAS magic-angle spinning
MeDOPE monomethyl-1,2-dioleoyl-sn-glycero-3- phosphoethanolamine
NBD (7-nitro-2-1,3-benzoxadiazol-4-yl)amino
NOE nuclear Overhauser effect
NOESY nuclear Overhauser enhancement spectroscopy
Phe phenylalanine
POPC 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
SDPE 1-stearoyl-2-docosahexaenoyl-sn-glycero-3- phosphoethanolamine
SDPC 1-stearoyl-2- docosahexaenoyl -sn-glycero-3- phosphocholine
SDPS 1-stearoyl-2-docosahexaenoyl-sn-glycero-3- phosphoserine
SOPC 1-stearoyl-2- oleoyl -sn-glycero-3-phosphocholine
Trp tryptophane
References
- Watts A. Solid-state NMR in drug design and discovery for membrane-embedded targets. Nature 2005;4:555-68.
- Drews J. Drug discovery: a historical perspective. Science 2000;287:1960-4.
- Kaiser ET, Kezdy FJ. Amphiphilic secondary structure: design of peptide hormones. Science 1984;223:249-55.
- Murray D, Ben-Tal N, Honig B, McLaughlin S. Electrostatic interaction of myristoylated proteins with membranes: simple physics, complicated biology. Structure 1997;5:985-9.
- Torres J, Stevens TJ, Samso M. Membrane proteins: the ‘Wild West’ of structural biology. Trends Biochem Sci 2003;28:137-44.
- http://blanco biomol uci edu/Membrane_Proteins_xtal html 2007;
- Feller SE, Huster D, Gawrisch K. Interpretation of NOESY cross-relaxation rates from molecular dynamics simulation of a lipid bilayer. J Am Chem Soc 1999;121:8963-4.
- Huster D. Investigations of the structure and dynamics of membrane-associated peptides by magic angle spinning NMR. Prog Nucl Magn Reson Spectrosc 2005;46:79-107.
- Pastor RW, Feller SE. Time scales of lipid dynamics and molecular dynamics. In: Merz KM, Roux B, editors. Biological Membanes. A molecular perspective from computation and experiment. Boston: Birkhäuser; 1996. p3–30.
- Huster D, Gawrisch K. New insights into biomembrane structure from two-dimensional nuclear overhauser enhancement spectroscopy. In: Katsaras J, Gutberlet T, editors. Lipid bilayers-structure and interactions. Berlin: Springer; 2000. p109–25.
- White SH, Wimley WC. Membrane protein folding and stability: physical principles. Annu Rev Biophys Biomol Struct 1999;28:319-65.
- White SH, Wimley WC. Hydrophobic interactions of peptides with membrane interfaces. Biochim Biophys Acta 1998;1376:339-52.
- White SH, Ladokhin AS, Jayasinghe S, Hristova K. How membranes shape protein structure. J Biol Chem 2001;276:32395-8.
- Wiener MC, White SH. Structure of a fluid dioleoylphosphatidyl-choline bilayer determined by joint refinement x-ray and neutron diffraction data III. Complete structure. Biophys J 1992;61:434-47.
- White SH, Wiener MC. The liquid-crystallographic structure of fluid lipid bilayer membranes. In: Merz KM, Roux B, editors. Biological Membranes. A Molecular Perspective from Computation and Experiment. Boston: Birkhäuser; 1996. p 127–44.
- Nagle JF, Tristram-Nagle S. Structure of lipid bilayers. Biochim Biophys Acta 2000;1469:159-95.
- Forbes J, Husted C, Oldfield E. High-field, high-resolution proton “magic-angle” sample-spinning nuclear magnetic resonance spectroscopic studies of gel and liquid crystalline lipid bilayers and the effects of cholesterol. J Am Chem Soc 1988;110:1059-65.
- Yeagle PL, Lee DA. Membrane protein structure. Biochim Biophys Acta 2002;1565:143.
- Luca S, Heise H, Lange A, Baldus M. Investigation of ligand-receptor systems by high-resolution solid-state NMR: recent progress and perspectives. Arch Pharm (Weinheim) 2005;338:217-28.
- Opella SJ, Marassi FM. Structure determination of membrane proteins by NMR spectroscopy. Chem Rev 2004;104:3587-606.
- Brown MF. Membrane structure and dynamics studied with NMR spectroscopy. In: Merz Jr. K, Roux B, editors. Biological Membranes. Boston: Birkhäuser; 1996. p 175–252.
- Auger M. Membrane structure and dynamics as viewed by solid-state NMR spectroscopy. Biophys Chem 1997;68:233-41.
- Gawrisch K, Eldho NV, Polozov IV. Novel NMR tools to study structure and dynamics of biomembranes. Chem Phys Lipids 2002;116:135-51.
- Lindblom G, Grobner G. NMR on lipid membranes and their proteins. Curr Opin Coll Int Sci 2006;11:24-9.
- Davis JH. The description of membrane lipid conformation, order and dynamics by 2H NMR. Biochim Biophys Acta 1983;737:117-71.
- Seelig J. Deuterium magnetic resonance: theory and application to lipid membranes. Q Rev Biophys 1977;10:353-418.
- Seelig J. 31P nuclear magnetic resonance and the head group structure of phospholipids in membranes. Biochim Biophys Acta 1978;515:105-40.
- Andrew ER, Bradbury A, Eades RG. Nuclear magnetic resonance spectra from a crystal rotated at high speed. Nature 1958;182:1659.
- Davis JH, Auger M, Hodges RS. High resolution 1H nuclear magnetic resonance of a transmembrane peptide. Biophys J 1995;69:1917-32.
- Jeener J, Meier BH, Bachmann P, Ernst RR. Investigation of exchange processes by two-dimensional NMR spectroscopy. J Chem Phys 1979;71:4546-53.
- Wagner G, Wüthrich K. Sequential resonance assignments in protein 1H nuclear magnetic resonance spectra. J Mol Biol 1982;155:347-66.
- Cavanagh J, Fairbrother WJ, Palmer AGI, Skelton NJ. Nuclear overhauser effect. In: Protein NMR Spectroscopy-Principles and Practice. San Diego: Academic Press; 1995. p 287–90.
- Wüthrich K. Nuclear overhauser enhancement (NOE) in biopolymers. In: NMR of Proteins and Nucleic Acids. New York: Wiley & Sons; 1986. p 93–113.
- Gawrisch K, Eldho NV, Polozov IV. Novel NMR tools to study structure and dynamics of biomembranes. Chem Phys Lipids 2002;116:135-51.
- Huster D, Arnold K, Gawrisch K. Investigation of lipid organization in biological membranes by two-dimensional nuclear overhauser enhancement spectroscopy. J Phys Chem B 1999;103:243-51.
- Yau WM, Gawrisch K. Lateral lipid diffusion dominates NOESY cross-relaxation in membranes. J Am Chem Soc 2000;122:3971-2.
- Macura S, Ernst RR. Elucidation of cross relaxation in liquids by two-dimensional NMR spectroscopy. Mol Phys 1980;41:95-117.
- Huster D, Arnold K, Gawrisch K. Influence of docosahexaenoic acid and cholesterol on lateral lipid organization in phospholipid mixtures. Biochemistry 1998;37:17299-308.
- Chen ZJ, Stark RE. Evaluating spin diffusion in MAS-NOESY spectra of phospholipid multibilayers. Solid State Nucl Magn Reson 1996;7:239-46.
- Forbes J, Bowers J, Shan X, Moran L, Oldfield E, Moscarello MA. Some new developments in solid-state nuclear magnetic resonance spectroscopic studies of lipids and biological membranes, including the effect of cholesterol in model and natural systems. J Chem Soc Faraday Trans 1 1988;84:3821-49.
- Huster D, Gawrisch K. NOESY NMR Crosspeaks between lipid headgroups and hydrocarbon chains: Spin diffusion or molecular disorder. J Am Chem Soc 1999;121:1992-3.
- Huster D, Jin AJ, Arnold K, Gawrisch K. Water permeability of polyunsaturated lipid membranes measured by 17O NMR. Biophys J 1997;73:855-64.
- Volke F, Pampel A. Membrane hydration and structure on a subnanometer scale as seen by high resolution solid state nuclear magnetic resonance: POPC and POPC/C12EO4 model membranes. Biophys J 1995;68:1960-5.
- Chen ZJ, Van Gorkom LC, Epand RM, Stark RE. Nuclear magnetic resonance studies of lipid hydration in monomethyldioleoyl-phosphatidylethanolamine dispersions. Biophys J 1996;70:1412-8.
- Zhou Z, Sayer BG, Hughes DW, Stark RE, Epand RM. Studies of phospholipid hydration by high-resolution magic-angle spinning nuclear magnetic resonance. Biophys J 1999;76:387-99.
- Gawrisch K, Gaede HC, Mihailescu M, White SH. Hydration of POPC bilayers studied by 1H-PFG-MAS-NOESY and neutron diffraction. Eur Biophys J 2007;36:281-91.
- Volke F, Eisenblätter S, Galle J, Klose G. Dynamic properties of water at phosphatidylcholine lipid-bilayer surfaces as seen be deuterium and pulsed field gradient proton NMR. Chem Phys Lipids 1994;70:121-31.
- Mitchell DC, Litman BJ. Effect of ethanol and osmotic stress on receptor conformation. Reduced water activity amplifies the effect of ethanol on metarhodopsin II formation. J Biol Chem 2000;275:5355-60.
- Mihic SJ, Ye Q, Wick MJ, Koltchine VV, Krasowski MD, Finn SE, et al. Sites of alcohol and volatile anaesthetic action on GABA(A) and glycine receptors. Nature 1997;389:385-9.
- Holte LL, Gawrisch K. Determining ethanol distribution in phospholipid multilayers with MAS- NOESY spectra. Biochemistry 1997;36:4669-74.
- Feller SE, Brown CA, Nizza DT, Gawrisch K. Nuclear overhauser enhancement spectroscopy cross-relaxation rates and ethanol distribution across membranes. Biophys J 2002;82:1396-404.
- Scheidt HA, Pampel A, Nissler L, Gebhardt R, Huster D. Investigation of the membrane localization and distribution of flavonoids by high-resolution magic angle spinning NMR spectroscopy. Biochim Biophys Acta 2004; 1663: 97–107.
- Siarheyeva A, Lopez JJ, Glaubitz C. Localization of multidrug transporter substrates within model membranes. Biochemistry 2006;45:6203-11.
- Dougherty DA. Cation-π Interactions in chemistry and biology: a new view of Benzene, Phe, Tyr, and Trp. Science 1996;271:163-8.
- Yau WM, Wimley WC, Gawrisch K, White SH. The preference of tryptophan for membrane interfaces. Biochemistry 1998;37:14713-8.
- Gaede HC, Yau WM, Gawrisch K. Electrostatic contributions to indole-lipid interactions. J Phys Chem B 2005;109:13014-23.
- Le GC, Seigneuret M. High-resolution mono- and multidimensional magic angle spinning 1H nuclear magnetic resonance of membrane peptides in nondeuterated lipid membranes and H2O. Biophys J 1996;71:2633-44.
- Zhang W, Crocker E, McLaughlin S, Smith SO. Binding of peptides with basic and aromatic residues to bilayer membranes: phenylalanine in the myristoylated alanine-rich C kinase substrate effector domain penetrates into the hydrophobic core of the bilayer. J Biol Chem 2003;278:21459-66.
- Epand RM, Sayer BG, Epand RF. Peptide-induced formation of cholesterol-rich domains. Biochemistry 2003;42:14677-89.
- Epand RF, Sayer BG, Epand RM. Induction of raft-like domains by a myristoylated NAP-22 peptide and its Tyr mutant. FEBS J 2005;272:1792-803.
- Epand RM, Sayer BG, Epand RF. Caveolin scaffolding region and cholesterol-rich domains in membranes. J Mol Biol 2005;345:339-50.
- Victor K, Jacob J, Cafiso DS. Interactions controlling the membrane binding of basic protein domains: phenylalanine and the attachment of the myristoylated alanine-rich C- kinase substrate protein to interfaces. Biochemistry 1999;38:12527-36.
- Simons K, Ikonen E. Functional rafts in cell membranes. Nature 1997;387:569-72.
- Ayala-Sanmartin J. Cholesterol enhances phospholipid binding and aggregation of annexins by their core domain. Biochem Biophys Res Commun 2001;283:72-9.
- Klein U, Gimpl G, Fahrenholz F. Alteration of the myometrial plasma membrane cholesterol content with beta-cyclodextrin modulates the binding affinity of the oxytocin receptor. Biochemistry 1995;34:13784-93.
- Albert AD, Young JE, Yeagle PL. Rhodopsin-cholesterol interactions in bovine rod outer segment disk membranes. Biochim Biophys Acta 1996;1285:47-55.
- Huster D, Kuhn K, Kadereit D, Waldmann H, Arnold K. 1H high-resolution magic angle spinning NMR spectroscopy for the investigation of a Ras lipopeptide in a lipid membrane. Angew Chem Int Ed Engl 2001;40:1056-8.
- Huster D, Vogel A, Katzka C, Scheidt HA, Binder H, Dante S, et al. Membrane insertion of a lipidated Ras peptide studied by FTIR, solid- state NMR, and neutron diffraction spectroscopy. J Am Chem Soc 2003;125:4070-9.
- Reuther G, Tan KT, Köhler J, Nowak C, Pampel A, Arnold K, et al. Structural model of the membrane-bound C terminus of lipid-modified human N-ras protein. Angew Chem Int Ed Engl 2006;45:5387-90.
- Huster D, Müller P, Arnold K, Herrmann A. Dynamics of membrane penetration of the fluorescent 7-nitrobenz-2-oxa- 1,3-diazol-4-yl (NBD) group attached to an acyl chain of phosphatidylcholine. Biophys J 2001;80:822-31.
- Huster D, Müller P, Arnold K, Herrmann A. The distribution of chain attached NBD in acidic membranes determined by 1H MAS NMR spectroscopy. Eur Biophys J 2003;32:47-54.
- Vogel A, Scheidt HA, Huster D. The distribution of lipid attached EPR probes in bilayers. Application to membrane protein topology. Biophys J 2003;85:1691-701.
- Scheidt HA, Müller P, Herrmann A, Huster D. The potential of fluorescent and spin labeled steroid analogs to mimic natural cholesterol. J Biol Chem 2003;278:45563-9.
- Scheidt HA, Flasche W, Cismas C, Rost M, Herrmann A, Liebscher J, et al. Design and application of lipophilic nucleosides as building blocks to obtain highly functional biological surfaces. J Phys Chem B 2004;108:16279-87.
- Cruciani O, Mannina L, Sobolev AP, Segre A, Luisi PL. Multilamellar liposomes formed by phosphatidyl nucleosides: an NMR-HR-MAS characterization. Langmuir 2004;20:1144-51.
- Kurz A, Bunge A, Windeck AK, Rost M, Flasche W, Arbuzova A, et al. Lipid-anchored oligonucleotides for stable double-helix formation in distinct membrane domains. Angew Chem Int Ed Engl 2006;45:4440-4.
- Bunge A, Kurz A, Windeck AK, Korte T, Flasche W, Liebscher J, et al. Lipophilic oligonucleotides spontaneously insert into lipid membranes, bind complementary DNA strands, and sequester into lipid-disordered domains. Langmuir 2007;23:4455-64.
- Petrache HI, Gouliaev N, Tristram-Nagle S, Zhang R, Suter RM, Nagle JF. Interbilayer interactions from high resolution x-ray scattering. Phys Rev E 1998;57:7014-24.
- Venable RM, Zhang Y, Hardy BJ, Pastor RW. Molecular dynamics simulations of a lipid bilayer and of hexadecane: an investigation of membrane fluidity. Science 1993;262:223-6.