
Role of oxidative stress in the apoptosis of hepatocellular carcinoma induced by combination of arsenic trioxide and ascorbic acid1
Introduction
Recently, the antineoplastic properties of arsenic trioxide (As2O3) have aroused wide interest. In vitro studies have shown that As2O3 induced the apoptosis of acute promyelocytic leukemia (APL) cell lines at micromolar concentrations[1]. These preclinical studies were confirmed by controlled clinical trials showing that treatment with As2O3 led to complete remission in APL patients[2]. More recently, studies have revealed that the apoptosis-inducing properties of As2O3 were not restricted to APL, and the viability of different types of tumor cells, including hepatocellular carcinoma cell lines, were shown to be altered in vitro by exposure to As2O3[3,4]. Key mediators of sensitivity to As2O3-induced apoptosis included intracellular glutathione (GSH) and reactive oxygen species (ROS). The loss of inner mitochondrial membrane potential is also an important step in As2O3-mediated cell apoptosis[5].
Ascorbic acid (AA), regarded as an anti-oxidant, has been found to have pro-oxidative properties which could enhance the sensitivity of myeloid and lymphoid malignant cells to As2O3 in vitro[6–8]. In a previous study, we found that AA could promote As2O3-induced apoptosis of human hepatocellular carcinoma cell line, HepG2, in vitro[9]. As ROS played a key role in AA combined with As2O3-induced apoptosis in other cell lines[6,7], we investigated ROS and downstream activation by ROS in the mechanisms of apoptosis induced by AA/As2O3 combination in HepG2 cells.
Materials and methods
Reagents Ascorbic acid, As2O3 and rhodamine 123 (RH123) were purchased from Sigma Chemical (USA) (chemical purification >99%). The 6-carboxy-2',7' dichloro-dihydrofluorescein diacetate (DCFH-DA) and N-acetyl-L-cysteine (NAC) were purchased from Beyotime Institute of Biotechnology; Annexin V-FITC Apoptosis Detection Kit was purchased from Alexis (USA); GSH, glutathione peroxidase (GPx) and superoxide dismutase (SOD) assay kits were purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China); antibodies to Bcl-2, Bax, and caspase-3 (p17) were obtained from BeiJing Zhongshan Golden Bridge Biotechnology Corporation (Beijing, China); and anti-β-actin antibody was purchased from Santa Cruz (USA).
Cell culture and drug treatment Human hepatocellular carcinoma cell line HepG2 was obtained from the China Center for Type Culture Collection. Cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (HyClone, USA) supplemented with 10% newborn calf serum (HyClone, USA) in a humidified atmosphere of 95% air and 5% CO2 at 37 °C in a carbon dioxide incubator (Shellab,USA). The cells in exponential growth state were divided into 4 groups: As2O3 treatment group, cells were incubated with 2.0 µmol/L As2O3 for 24 h; AA treatment group, cells were treated with 100 µmol/L AA for 24 h; combined treatment group, cells were cultured in 2.0 µmol/L As2O3 combined with 100 µmol/L AA for 24 h; control group, cells were incubated for 24 h with medium alone. In the experiments on apoptosis rate and ROS level, the above 4 groups were treated with or without 10 mmol/L NAC for 24 h.
Detection of intracellular ROS Intracellular ROS was detected by means of an oxidation-sensitive fluorescent probe (DCFH-DA). After treatment with As2O3 2.0 µmol/L and/or AA 100 µmol/L, with or without NAC 10 mmol/L for 24 h, cells were washed twice in phosphate-buffered saline (PBS). They were then incubated with 10 µmol/L DCFH-DA at 37 ºC for 20 min according to the manufacturer’s instructions. DCFH-DA was deacetylated intracellularly by nonspecific esterase, which was further oxidized by ROS to the fluorescent compound 2,7-dichlorofluorescein (DCF). DCF fluorescence was detected by FACScan flow cytometer (Becton Dickinson). For each sample 10 000 events were collected.
Intracellular GSH, and GPx, SOD activity assay The GSH activity was measured by reacting on DTNB (5,5'-DITHIO-BIS(2-NITROBENZOIC ACID) to form a yellow substance, which can be determined by colorimetry. The GPx activity was detected by the oxidizing speed of the GSH, which can be expressed by the GSH reduction in a certain time. One unit of GPx activity was defined as 1 µmol/L GSH oxidized to GSSG (glutathione disulphide) per milligram of protein per minute. The SOD activity was determined by hydroxylamine assay-developed from xanthine oxidase assay, which was presented as units per milligram of protein. The 4 groups of treated cells mentioned in “cell culture and drug treatment” were washed twice and then resuspended in PBS, sonicated for 30 s on ice, and centrifuged at 1000×g for 15 min according to the manufacturer’s instructions. The supernatants were subjected to intracellular GSH, and GPx, SOD activity assays, which were determined with commercial kits from Jiancheng Bioengineering Institute.
Flow cytometry analysis of mitochondrial membrane potential RH123, the anion dye, could enter the mitochondrial matrix and cause photoluminescent quenching dependent on mitochondrial transmembrane potential. If mitochodrial membrane potential depleted, the RH123 could be released from the mitochondria and produce fluorescence. The mean fluorescence intensity of RH123 was measured to determine the loss of mitochondrial membrane potential. Treated cells were collected and resuspended at a concentration of 1×105/mL in PBS, which contained 1 µmol/L RH123 and incubated at 37 ºC for 30 min. Samples were analyzed by FACScan flow cytometer (Becton Dickinson, USA) with an excitation wavelength of 488 nm and an emission wavelength of 525 nm. Fluorescent signal intensity was recorded and analyzed by CellQuest software (Becton Dickinson). For each sample 10 000 events were collected[10].
Western blotting analysis of Bcl-2, Bax and p17 subunit of caspase-3 expression Approximately 5×106 treated cells were washed with cold PBS (containing PMSF, phenyl-methylsulfonyl fluoride) three times, and collected by scraping. Collected cells were solubilized in 150 µL ice-cold lyse buffer, and then sonicated. The cell-free supernatants were recovered by centrifugation at 12 000×g for 25 min at 4 °C. Loading buffer was added to each supernatant, which was subsequently boiled for 10 min. Then 20 µL lysate was electrophoresed on a 10% sodium dodecyl sulfate (SDS) polyacrylamide gel. Proteins were then transferred onto nitrocellulose membrane. After blocking of the membrane with 5% non-fat dry milk in Tris-buffered saline containing 0.1% Tween-20 for 1 h at 25 °C, the blots were incubated for 12 h at 4°C with the appropriate dilution of the studied antibody (anti-Bcl-2, anti-Bax and anti-caspase-3 p17 antibody was used at 1:500 dilution; anti-β-actin antibody was used at 1:3000 dilution). After washing, the blots were incubated with peroxidase-conjugated secondary antibody (1:2500) in the second reaction. Binding of antibodies was detected by chemiluminescence staining using the ECL detection kit (Amersham) and quantified by densitometry. Comparable loading of proteins on the gel was verified by re-probing the blots with an antibody specific for the housekeeping gene product β-actin. The optical density scores of bands were calculated to analyze the relative content of Bcl-2, Bax, and caspase-3 after normalization with β-actin values in the same lane.
Apoptosis assessment by Annexin V-FITC and propidium iodide staining Cell apoptosis was measured by Annexin V-FITC and propidium iodide (PI) staining. Cells were treated with As2O3 2.0 µmol/L and/or AA 100 µmol/L, with or without NAC 10 mmol/L for 24 h, and then washed twice in 4 °C PBS and resuspended in binding buffer at a cell density of 1×106/mL. Cells were then stained with 5 µL Annexin V-FITC and 10 µL PI (20 µg/mL) according to the manufacturer’s instructions. They were then incubated in the dark at 25 °C for 15 min. Samples were acquired on a FACScan flow cytometer (Becton Dickinson) and analyzed with Cellquest software (Becton Dickinson); 10 000 cells were analyzed.
Statistical analyses All values were presented as the mean±SD. Statistical analyses were performed by using SPSS 11.0 software. Representative data were analyzed for statistical significance by one-way ANOVA. A P-value of less than 0.05 was considered as statistically significant.
Results
Effects of AA and As2O3 on intracellular ROS level in HepG2 cells Because the production of ROS is a critical contributor to As2O3-induced apoptosis[5], the level of ROS was measured to test whether oxidative stress played an important role in AA potentiated As2O3-induced apoptosis. Exposure of HepG2 cells to 2.0 µmol/L As2O3 plus 100 µmol/L AA led to a remarkable increase in intracellular ROS compared with As2O3 alone, with increased ROS level represented by the DCF intensity from 127.61±5.12 to 152.60±5.88 (Figure 1, Table 1).
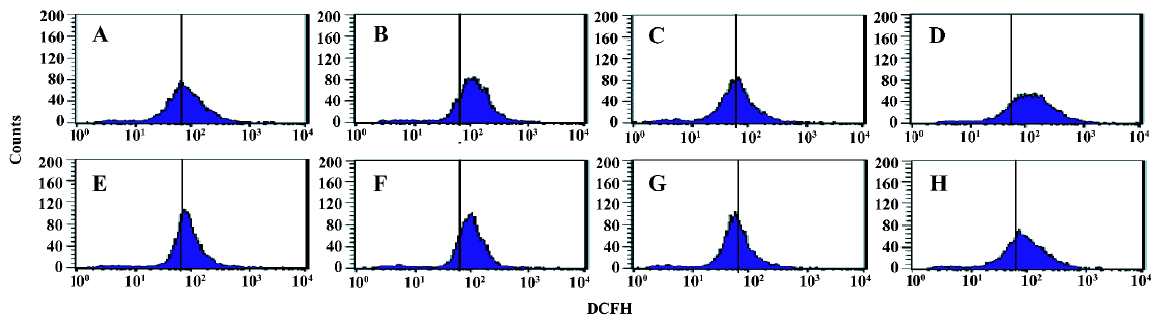
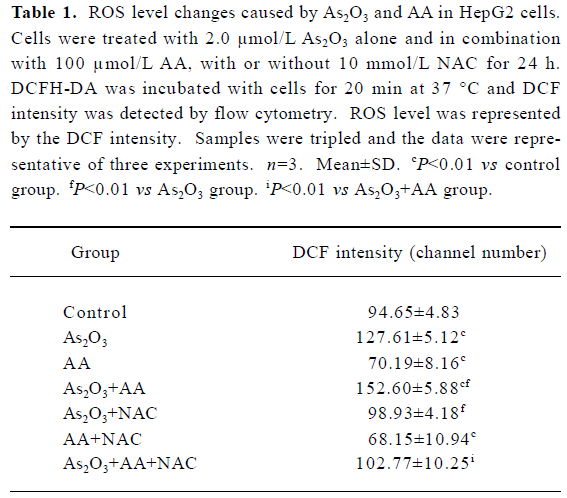
Full table
Effects of AA and As2O3 on intracellular GSH level in HepG2 cells A remarkable depletion in cellular GSH was found after AA or As2O3 exposure. A higher decrease in GSH was observed in the combined treatment than As2O3 treatment (Table 2). As GSH was recognized as the predictor for response to As2O3 treatment[5], the results suggested that AA potentiated As2O3-induced apoptosis by depleting intracellular GSH.
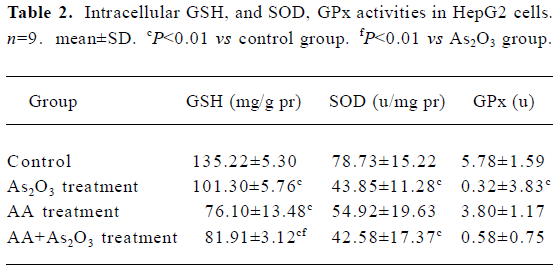
Full table
Effects of AA and As2O3 on intracellular GPx and SOD activity In this research, significant reductions in GPx and SOD activity were observed after As2O3/AA exposure (Table 2). These results also proved that oxidative insults occurred in the combined treatment, because GPx and SOD were recognized as the most important antioxidant members. However, SOD and GPx depletion were found to be equal to As2O3 treated alone (P>0.05).
Effects of AA and As2O3 on mitochondria membrane potential ROS can result in free radical attack of membrane phospholipids, leading to a loss of mitochondrial membrane potential. Mitochondria depolarization was considered as an irreversible step in the apoptosis process[11]. Therefore, the ability of HepG2 cell mitochondria to maintain membrane potential in the four groups was measured by using flow cytometry techniques. As2O3 treatment caused depolarization of the inner mitochondrial membrane, while AA alone had no significant effect on mitochondrial membrane potential (Figure 2). The combination of As2O3 and AA increased the depolarization of the inner mitochondrial membrane compared to As2O3 treatment alone (Figure 2).
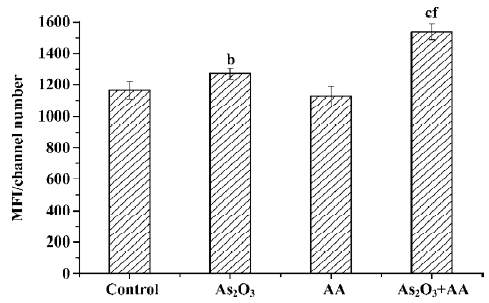
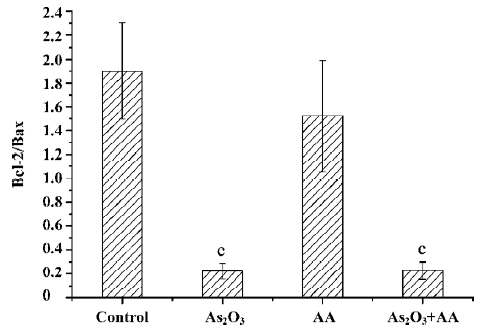
Effects of AA and As2O3 on the ratio of Bcl-2/Bax Proto-oncogene Bcl-2 appeared to function by inhibiting the mitochondria depolarization. Conversely, Bax induced mitochondria depolarization. The ratio of Bcl-2 to Bax has been reported to be correlated with apoptosis in cancer cells. The anti-apoptotic Bcl-2 protein bound a pro-apoptotic Bax protein, and if Bcl-2 was in excess, all available Bax was bound and apoptosis was blocked. If Bax was in excess, all Bcl-2 was bound and the cell proceeded to die[12]. Our results showed that there was an approximate 9-fold decrease in the ratio of Bcl-2/Bax expression in the As2O3 treatment and combined treatment compared with the control, but there was no significant alteration between As2O3 treatment and the combined treatment (Figures 3,4,5). Therefore, we could infer that AA potentiated As2O3-induced apoptosis was in a Bcl-2 and Bax independent manner.
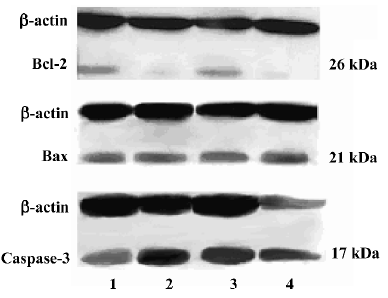
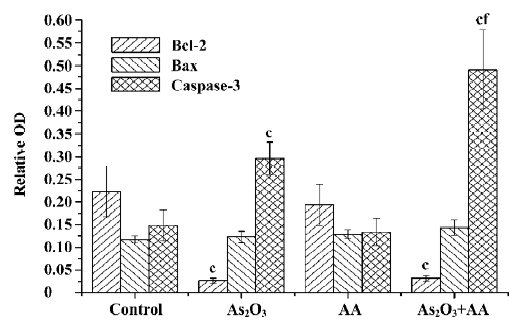
Effects of AA and As2O3 on activation of caspase-3 With the mitochondrial membrane’s potential depletion by ROS, the permeability transition pore (PTP) opened and intermembrane proteins were released out of the mitochondria, which in turn activated a downstream executive caspase-3[13]. Caspase-3 is expressed in cells as an inactive 32 kDa precursor from which the 17 kDa (p17) and 11 kDa (p11) subunits of the mature caspase-3 are proteolytically generated during apoptosis. The active caspase-3 enzyme is a heterodimer composed of two p17 and two p11 subunits. In the present study, a marked increase of caspase-3 p17-activated caspase-3 subunit in As2O3 or combined treatment was observed. Furthermore, a significant increase in the combined treatment group compared with the As2O3 treatment group was found (Figures 3 and 4).
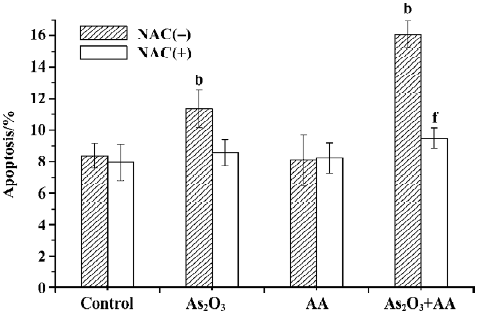
NAC attenuates As2O3 and AA-mediated ROS increase and cell apoptosis In our previous studies, AA/As2O3 combined treatment could increase the apoptosis rate significantly compared to As2O3 treatment alone[9]. Similarly, in this test, untreated cultures contained 7.89%±0.98% apoptotic cells. However, if the cells were treated with 2.0 µmol/L As2O3 and 100 µmol/L AA in combination, an almost 2-fold increase to 15.60%±1.14% apoptotic cells was observed. In order to find out the relationship between ROS and the apoptosis effect of combined treatment, antioxidant NAC was used to scavenge intracellular ROS. The results showed that NAC abolished the apoptosis effect of As2O3 and AA-combined treatment, with the apoptosis rate of 9.48%±0.67% (Figure 6), while attenuating combinative treatment mediated ROS elevation (Figure 1, Table 1). These results suggested that ROS played a very important role in the enhancement effect of AA on the apoptosis of HepG2 cells induced by As2O3.
Discussion
ROS played a very important role in apoptosis induction under both physiological and pathological conditions. Interestingly, mitochondria were both the source and target of ROS. ROS, which was predominantly produced in the mitochondria, led to the free radical attack of membrane phospholipids and loss of mitochondrial membrane potential, which caused the intermembrane proteins, such as cytochrome c, to be released out of the mitochondria and ultimately triggered caspase-3 activation. Caspase-3 activation led to DNA breakage, nuclear chromatin condensation, and cell apoptosis[5,11,13,17]. Anti-apoptotic Bcl-2 and pro-apoptotic Bax, which resided upstream of mitochondria, focused much of their efforts at the level of mitochondria. Bcl-2 appeared to inhibit the mitochondria depolarization and ROS production, while Bax induced mitochondria depolarization and ROS production[12].
Oxidative damage has been postulated to play the key role in the apoptosis process induced by As2O3[14–16,23,24]. The evidence was listed as follows: first, As2O3 disturbed natural oxidation and reduction equilibria through various mechanisms involved in complex redox reactions with endogenous oxidants and cellular antioxidant systems; second, As2O3, by oxidizing thiols, acted directly on mitochondria to destroy the mitochondrial inner transmembrane potential, then promoted ROS production; and third, As2O3, by downregulating Bcl-2 expression, resulted in mitochondria depolarization and ROS production[1,22]. A major mechanism for detoxification of radicals in mammalian cells is the breakdown of ROS by scavenger, such as GPx, and SOD. The former could specifically catalyse the GSH to reduce the hydroperoxide[14], and the latter could clear the superoxide anion free radical.
AA, which was regarded as an anti-oxidant, was found to have pro-oxidative properties. In the plasma, AA was rapidly oxidized to dehydroascorbic acid (DHA), which was then transported into the cell. AA was then regenerated from DHA via glutaredoxin, which converted GSH to GSH disulfide. Recent evidence showed that AA enhanced the sensitivity of myeloid and lymphoid malignant cells to As2O3 in vitro through the oxidative pathway[6–8] and the increased level of ROS was the result of GSH depletion. GSH was a main nonprotein antioxidant in the cell, and it could clear away the superoxide anion free radical and provide electrons for enzymes such as GPx, which reduce H2O2 to H2O. Additionally, GSH could combine with As2O3 directly, forming a transient As(GS)3 molecule, which was more easily excreted through drug efflux pumps[18–21].
In the present study, ROS was found to be a critical contributor to the potentiating effect of AA on As2O3-induced apoptosis in HepG2 cells. A higher level of ROS production, much more depletion of antioxidant enzymes, and the activation of ROS downstream, such as mitochondria depolarization and caspase-3 activation, were detected in the combined treatment as compared to the As2O3 treatment alone. This suggests that AA potentiated As2O3 -induced apoptosis in HepG2 cells through the oxidative pathway. More importantly, As2O3 and AA mediated cell apoptosis was abrogated in the presence of an antioxidant, NAC, which at the same time attenuated ROS elevation caused by the As2O3 and AA combination. Additionally, we found that AA enhanced As2O3-induced ROS production was related to GSH depletion and was independent of Bcl-2 and Bax, although As2O3 alone has been shown to cause downregulation of the ratio of Bcl-2 to Bax protein in HepG2 cells.
In conclusion, AA, by depleting intracellular GSH, potentiated As2O3 mediated ROS production, which enhanced the loss of mitochondrial membrane potential and activation of caspase-3, thus potentiating apoptosis in HepG2 cells induced by As2O3.
References
- Chen GQ, Zhu J, Shi XG, Ni JH, Zhong HJ, Si GY, et al. In vitro studies on cellular and molecular mechanisms of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia: As2O3 induces NB4 cell apoptosis with downregulation of Bcl-2 expression and modulation of PML-RAR alpha/PML proteins. Blood 1996;88:1052-61.
- Soignet SL, Maslak P, Wang ZG, Jhanwar S, Calleja E, Dardashti LJ, et al. Complete remission after treatment of acute promyelocytic leukemia with arsenic trioxide. N Engl J Med 1998;339:1341-8.
- Michel L, Dupuy A, Jean-Louis F, Sors A, Poupon J, Viguier M, et al. Arsenic trioxide induces apoptosis of cutaneous T cell lymphoma cells: evidence for a partially caspase-independent pathway and potentiation by ascorbic acid (vitamin C). J Investigative Dermatol 2003;121:881-93.
- Oketani M, Kohara K, Tuvdendorj D, Ishitsuka K, Komorizono Y, Ishibashi K, et al. Inhibition by arsenic trioxide of human hepatoma cell growth. Cancer Lett 2002;183:147-53.
- Miller WH Jr, Schipper HM, Lee JS, Singer J, Waxman S. Mechanisms of action of arsenic trioxide. Cancer Res 2002;62:3893-903.
- Grad JM, Bahlis NJ, Reis I, Oshiro MM, Dalton WS, Boise LH. Ascorbic acid enhances arsenic trioxide-induced cytotoxicity in multiple myeloma cells. Blood 2001;98:805-13.
- Bahlis NJ, McCafferty-Grad J, Jordan-McMurry I, Neil J, Reis I, Kharfan-Dabaja M, et al. Feasibility and correlates of arsenic trioxide combined with ascorbic acid-mediated depletion of intracellular glutathione for the treatment of relapsed/refractory multiple myeloma. Clin Cancer Res 2002;8:3658-68.
- Bachleitner-Hofmann T, Gisslinger B, Grumbeck E, Gisslinger H. Arsenic trioxide and ascorbic acid: synergy with potential implications for the treatment of acute myeloid leukaemia? Br J Haematol 2001;112:783-6.
- Li Y, Li JJ, Tang Q, Fu Q, Hu BR, Xiang JZ. Study on the apopto-sis of human hepatocarcinoma cells by combined vitamin c with arsenic trioxide. Herald Med 2006;1:3-5.
- Ludovico P, Sansonetty F, Corte-Real M. Assessment of mitochondrial membrane potential in yeast cell populations by flow cytometry. Microbiology 2001;147:3335-43.
- Pelicano H, Feng L, Zhou Y, Carew JS, Hileman EO, Plunkett W, et al. Inhibition of mitochondrial respiration: a novel strategy to enhance drug-induced apoptosis in human leukemia cells by a reactive oxygen species-mediated mechanism. J Biol Chem 2003;278:37832-9.
- Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes Dev 1999;13:1899-911.
- Bras M, Queenan B, Susin SA. Programmed cell death via mitochondria: Different modes of dying. Biochemistry 2005;70:231-9.
- Jing Y, Dai J, Chalmers-Redman RM, Tatton WG, Waxman S. Arsenic trioxide selectively induces acute promyelocytic leukemia cell apoptosis via a hydrogen peroxide-dependent pathway. Blood 1999;94:2102-11.
- Liu L, Trimarchi JR, Navarro P, Blasco MA, Keefe DL. Oxidative stress contributes to arsenic-induced telomere attrition, chromosome instability, and apoptosis. J Biol Chem 2003;278:31998-2004.
- Yang J, Li H, Chen YY, Wang XJ, Shi GY, Hu QS, et al. Anthraquinones sensitize tumor cells to arsenic cytotoxicity in vitro and in vivo via reactive oxygen species-mediated dual regulation of apoptosis. Free Radic Biol Med 2004;37:2027-41.
- Kroemer G, de The H. Arsenic trioxide, a novel mitochondriotoxic anticancer agent? J Natl Cancer Inst 1999;91:743.
- Dai J, Weinberg RS, Waxman S, Jing Y. Malignant cells can be sensitized to undergo growth inhibition and apoptosis by arsenic trioxide through modulation of the glutathione redox system. Blood 1999;93:268-77.
- Kito M, Akao Y, Ohishi N, Yagi K, Nozawa Y. Arsenic trioxide-induced apoptosis and its enhancement by buthionine sulfoximine in hepatocellular carcinoma cell lines. Biochem Biophys Res Commun 2002;291:861-7.
- Gregus Z, Gyurasics A. Role of glutathione in the biliary excretion of the arsenical drugs trimelarsan and melarsoprol. Biochem Pharmacol 2000;59:1375-85.
- May JM, Qu Z, Li X. Requirement for GSH in recycling of ascorbic acid in endothelial cells. Biochem Pharmacol 2001;62:873-81.
- Mahieux R, Pise-Masison C, Gessain A, Brady JN, Olivier R, Perret E, et al. Arsenic trioxide induces apoptosis in human T-cell leukemia virus type 1- and type 2-infected cells by a caspase-3-dependent mechanism involving Bcl-2 cleavage. Blood 2001;98:3762-9.
- Yi J, Yang J, He R, Gao F, Sang H, Tang X, et al. Emodin enhances arsenic trioxide-induced apoptosis via generation of reactive oxygen species and inhibition of survival signaling. Cancer Res 2004;64:108-16.
- Sturlan S, Baumgartner M, Roth E, Bachleitner-Hofmann T. Docosahexaenoic acid enhances arsenic trioxide–mediated apoptosis in arsenic trioxide-resistant HL-60 cells. Blood 2003;101:4990-7.