
Influence of connective tissue growth factor antisense oligonucleotide on angiotensin II-induced epithelial mesenchymal transition in HK2 cells1
Introduction
At present, there is increasing evidence suggesting that activation of the intrarenal rennin-angiotensin system (RAS) might play a key role in controlling the progression of renal fibrosis[1]. Angiotensin II (Ang II), the main peptide of the rennin-angiotensin system, is a true cytokine that regulates cell growth, inflammation, and fibrosis, contributing to the progression of renal disease. Blocking the effects of Ang II either by Ang II-converting enzyme inhibitors (ACEI) or Ang II receptor antagonists (ARA) has been demonstrated to successfully prevent renal fibrogenesis by blocking its hemodynamic and nonhemodynamic action[2,3]. However, although a number of recent studies have demonstrated that Ang II might be a major contributor in regulating renal cell transdifferentiation, its mechanism is not fully understood.
Connective tissue growth factor (CTGF), a member of the CCN (CTGF, cyr61, nov) immediate early gene family of proteins, is a 38-kD, cysteine-rich, heparin-binding protein. CTGF has been implicated in mitogenic activity, cell migration, fibroblast proliferation, cellular adhesion, angiogenesis and ECM (extracellular matrix) synthesis. Increased expression of CTGF has been found in inflammatory bowel disease, skin lesions, renal fibrosis, liver fibrosis and idiopathic pulmonary fibrosis, suggesting that CTGF may be involved in the pathogenesis of fibrosis[4]. Furthermore, urinary CTGF levels are elevated in many different renal fibrotic disorders, including diabetic nephropathy, IgA nephropathy, focal segmental glomerulosclerosis, crescentic nephritis and lupus nephritis[5]. It is accordingly thought that CTGF can drive glomerulosclerosis and tubulointerstitial fibrosis in a variety of renal diseases.
We have demonstrated in a previous study that in vitro expression of CTGF correlates with the early enlargement of glomeruli in diabetic rats, which can be significantly prevented by treatment with an Ang II receptor antagonist irbesartan. Simultaneously, irbesartan can also inhibit the over expression of CTGF in diabetic rats[6]. In in vivo study, we have found that Ang II stimulated the tubular epithelial cell hypertrophy and CTGF expression, which could be significantly inhibited by cotreatment with anti-CTGF antibody[7].
Tubulointerstitial fibrosis is considered to be the final common pathway leading to end-stage renal failure, irrespective of the nature of the initial renal injury. Recent studies have demonstrated that tubular epithelial mesenchymal transition (EMT) is an important precursor to the development of tubulointerstitial fibrosis, and is detrimental to the kidney in the long term[8,9]. Proximal tubular epithelial cells (PTEC) are increasingly being recognized for playing a central role in renal tubulointerstitial fibrosis. Obviously, it is quite important to understand the exact mechanisms of tubular epithelial cell EMT that will provide a new strategy for early prevention of renal fibrosis.
Although Ang II and CTGF are considered to be important mediators in the process of renal fibrosis, especially in EMT, the relationship between CTGF and Ang II in mediating EMT need to be further clarified. Our present study was an investigation of the influence of CTGF-antisense oligonucleotide (CTGF-AS) on Ang II-induced tubular cell EMT, thus identifying the role CTGF in the process of Ang II-induced EMT.
Materials and methods
Cell culture HK2 cells (human proximal tubular cells immortalized by transduction with human papilloma virus E6/E7 gene) were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 1000 mg/L glucose, 100 U/mL penicillin, 100 µg/mL streptomycin, 2 mmol/L supplemental glutamine and 10% heat-inactivated fetal calf serum (Sijiqing, China) at 37 ºC in 5% CO2. Cultural medium was changed every day and cells were passaged every 48–72 h by trypsin.
Reagents Ang II was purchased from Sigma (Saint Louis Mo, USA). 16-mer CTGF antisense phosphorothioate oligonucleotide (5'-TACTGGCGGCGGTCAT-3') containing the initial ATG translation start site was synthesized and purchased from China Shanghai Genebase (Shanghai, China). An oligonucleotide containing a scrambled nucleotide sequence (5'-GGTCTAGCTTGCGGAC-3') was used as the control[10]. The synthetic oligonucleotides were added directly to the cell culture medium (final concentration, 20 µg/mL).
RT-PCR analysis To gain information as to whether HK2 cells express CTGF and to observe the effect of CTGF-AS on Ang II-induced expression of CTGF mRNA, cDNA amplification was performed after reverse transcription of RNA. The HK2 cell line was planted in a six-well plate at a density of 1×10 5 cells/well. When reaching 90% confluence, the cells were incubated with serum-free DMEM for 24 h of quiescence. Cells were then incubated separately with serum-free medium, Ang II (1×10-7 mol/L), Ang II (1×10-7 mol/L) plus CTGF-AS (20 µg/mL), Ang II (1×10-7 mol/L) plus scrambled oligonucleotide (20 µg/mL) for 48 h. Total RNA was isolated from cultured cells by the TRIZOL method. RNA was quantitated by UV absorption measurement at 260 nm. The absorption ratio of OD260/OD280 was between 1.8 and 2.0. Reverse transcript polymase chain reaction (RT-PCR) was performed using access RT-PCR kit (Promega, Madison, WI, USA). The primer sequences were as follows[11]:
CTGF: forward: 5'-GAGGAAAACATTAAGAAGGGCAAA-3';
reverse: 5'-CGGCACAGGTCTTGATGA-3';
GAPDH: forward: 5'-CTCAGACACCATGGGGAAGGTGA-3';
reverse: 5'-ATGATCTTGAGGCTGTTGTCATA-3'.
Total RNA (1 µg) was added into each response system. The parameters of RT-PCR were 45 min at 48 ºC, 2 min at 94 ºC, then 40 cycles (94 ºC 1 min, 50 ºC 1 min, 68 ºC 2 min), and an additional extension step for 7 min at 68 ºC as the last cycle. Ten microliters of the reaction products were electrophoresed in a 2% agarose gel containing 0.5 µg/mL ethidium bromide. It was then scanned by the IMAGE MASTER VDS and analyzed with Total Lab software. Results were expressed as the ratio of CTGF mRNA to GAPDH mRNA. RT-PCR with separate stimulation of cells was independently performed three times.
Indirect-immunofluorescence The bacteria-free coverglasses were dealed with PolyLysine, and then placed on 24-well plates (NUNC). The HK2 cell line was plated in 24-well plates (1×105 per well). When cells reached 80% confluence, they were rested in serum-free medium for 24 h. They were then treated separately with serum-free medium, Ang II (1×10-7 mol/L), Ang II (1×10-7 mol/L) plus CTGF-AS (20 µg/mL), Ang II (1×10-7 mol/L) and scrambled oligonucleotide (20 µg/mL), CTGF-AS (20 µg/mL) and scrambled oligonucleotide (20 µg/mL) for 48 h. The coverglasses of confluent culture were removed and immunofluorescence was carried out. The cells were fixed and permeabilized. Nonspecific sites were then blocked. The primary antibodies [goat anti-human CTGF (1:100), R&D Systems] were incubated with the cells overnight at 4 ºC in a humidified chamber. Washed cells were incubated with the appropriate secondary antibody-conjugate (1:100) for 1 h at room temperature. The coverslips were washed. The cells were inspected using a Confocal microscope (ZEISS LSM510, Oberkochen, Germany) and were analyzed by LSM510 Version 2.3 software, provided with the machine.
Cellular ultrastructure Cells were seeded in 100-mL culture bottles in equal numbers. After the cells reached 80% confluence, they were rested in serum-free medium for 24 h. They were then treated separately with serum-free medium, Ang II (1×10-7 mol/L), Ang II (1×10-7 mol/L) plus CTGF-AS (20 µg/mL) for 48 h/96 h. Cells were trypsinized, centrifuged, washed and collected. They were then fixed at 4 ºC in 2% glutaraldehyde for 2 h. After being dehydrated, soaked, embedded, sectioned and stained they were examined with a HITACHI H600 electron microscope.
Immunocytochemistry The bacteria-free coverglasses were dealed with PolyLysine overnight, and then placed on 24-well plates. The HK2 cell line was plated on 24-well plates (1×105 per well). When cells reached 80% confluence, they were rested in serum-free medium for 24 h. Ang II (1×10-7 mol/L) was added to the medium at various times (0, 24, 48, 72, and 96 h). Meanwhile, the effect of Ang II (1×10-7 mol/L) and CTGF-AS (20 µg/mL) at 72 h was observed. Staining for α-smooth muscle actin (α-SMA) was performed using a monoclonal mouse anti-human α-SMA antibody [monoclonal mouse anti-human α-SMA (1:50), Beijing Zhongshan Biotechnology, China] in appropriate buffer according to anti-mouse/rabbit UltraSensitive S-P kit instructions. The coverglasses were stained with AEC (3-amino-9-ethyl carbazole) and counterstained with hematoxylin then cleared. The cells were divided into four groups on the basis of the stain intensity, which includes negative (–), weakly positive (+), positive (++), and strongly positive (+++). The cells per coverglass were counted.
Statistical analysis All data are expressed as mean±SD. Multiple groups of values were compared using one-way analysis of variance (ANOVA). Data were analyzed by SPSS 10.0 programs for Windows. The data of immunocytochemistry were analyzed by rank sum test. A P value of less than 0.05 was considered statistically significant.
Results
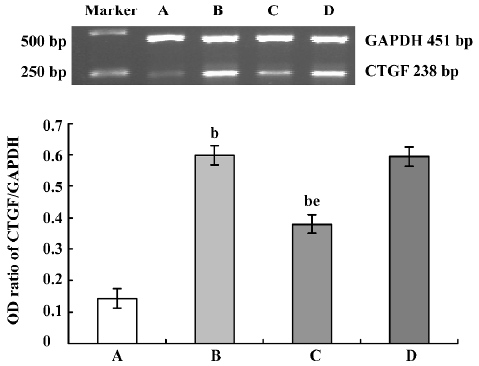
Influence of CTGF-AS on Ang II-induced expression of CTGF mRNA in HK2 cells In response to treatment with Ang II (1×10-7 mol/L), CTGF mRNA expression increased significantly (OD ratio: control group: 0.144±0.024 vs Ang II group: 0.598±0.033, P<0.01), which was markedly attenuated in the presence of CTGF-AS (P<0.01). The scrambled oligonucleotide had no significant effect on Ang II-induced CTGF mRNA expression (P>0.05) (Figure 1).
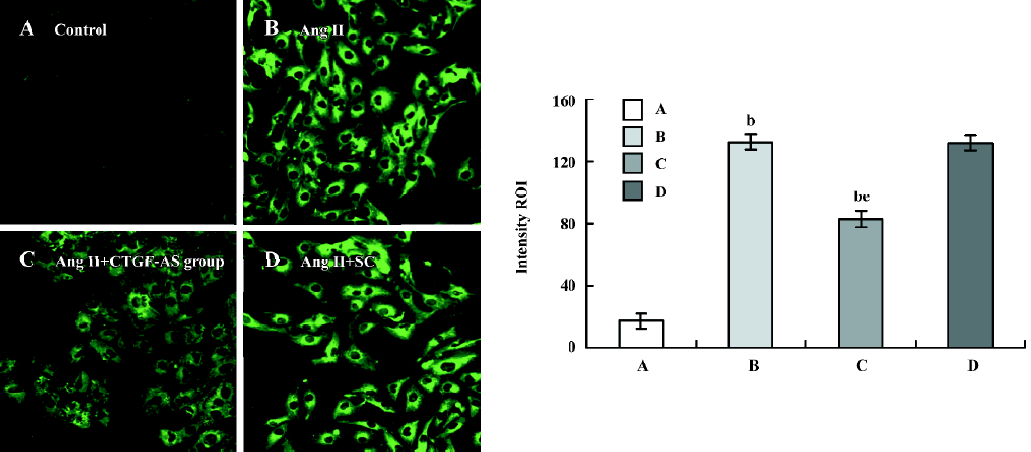
Influence of CTGF-AS on Ang II-induced expression of CTGF protein in HK2 cells The expression of CTGF protein in tubular cells was measured using the Intensity ROI by Confocal microscope. The stimulation of HK2 cells with Ang II (1×10-7 mol/L) for 48 h resulted in a significant increase in the synthesis of CTGF protein (Intensity ROI: control group: 17.41±5.04 vs Ang II group: 132.45±5.10, P<0.01). In addition, this change was indeed abolished by cotreatment with CTGF-AS (AngII group: 132.45±5.10 vs Ang II+CTGF-AS group: 82.77±5.09, P<0.01). The scrambled oligonucleotide had no significant effect on Ang II-induced CTGF protein expression (P>0.05) (Figure 2).
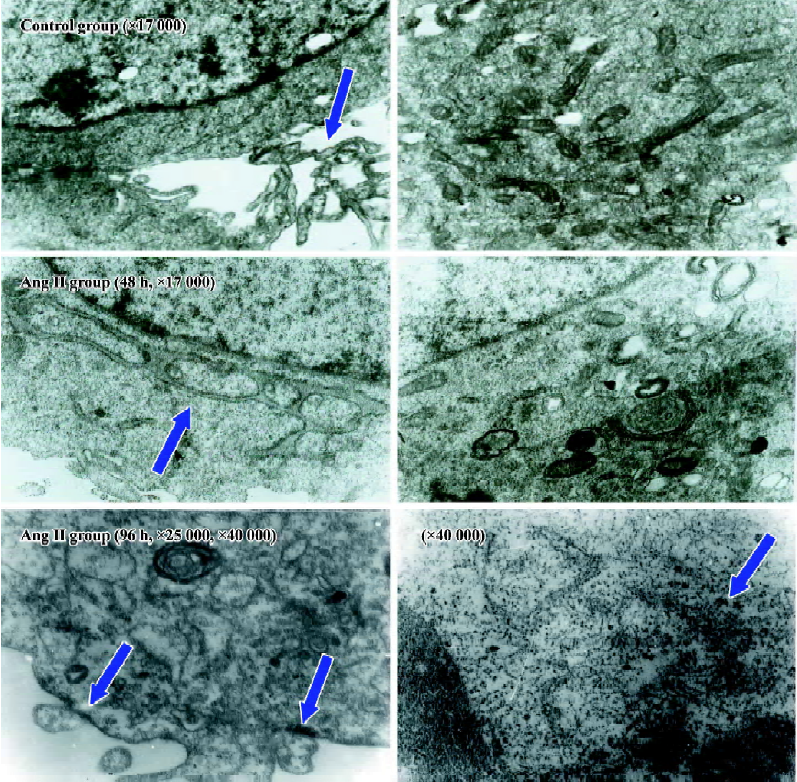
Influence of CTGF-AS on Ang II-induced alterations of cellular ultrastructure in HK2 cells The cellular ultrastructure of control group presented with rich microvilli and the mitochondria. After stimulating HK2 cells with Ang II for 48 h, the cellular ultrastructure showed the microvilli and the mitochondria decreased, the rough endoplasmic reticulum and the Golgi apparatus increased. After stimulating HK2 cells with Ang II for 96 h, the cellular ultrastructure showed the cellular membrane had pykno-architecture and the filament architecture were dispersed in the cytoplasm. These effects could be partially abolished by treatment with CTGF-AS (Figure 3).
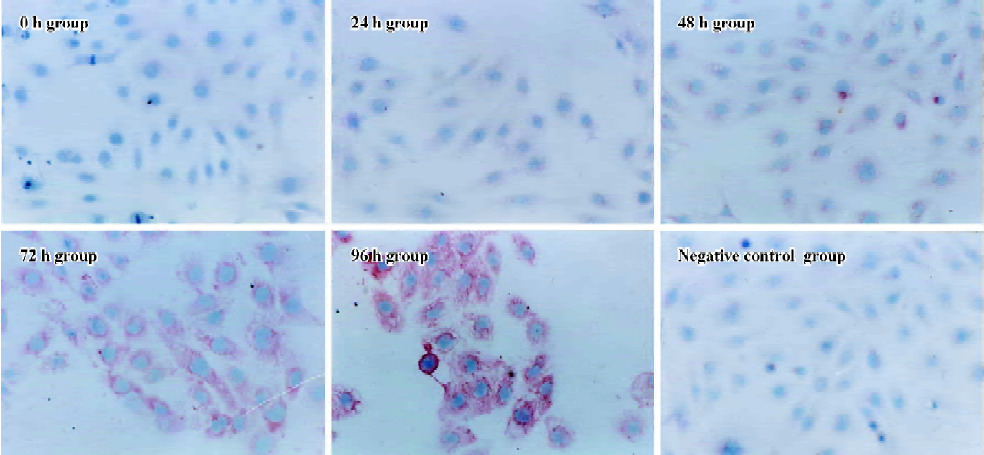
Influence of CTGF-AS on AngII-induced expression of α-SMA protein in HK2 cells α-SMA protein levels were assessed by immunohistochemistry. After treating HK2 with 1×10-7 mol/L Ang II at different time points, it was shown that no α-SMA immunostaining was observed in 0 h group. In response to Ang II, α-SMA was initially detectable at 48 h, a significant increase in positive α-SMA immunostaining was detectable at 72 h, and a progressive increase occurred over the 96 h of study. The synthesized level of α-SMA reached a peak at 96 h. It was found that Ang II time-dependently induced synthesis of α-SMA in the HK2 cells (Figure 4, Table 1).
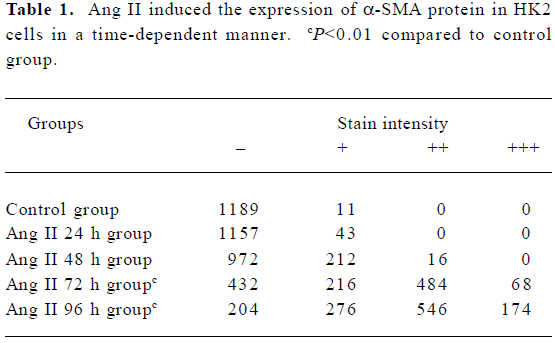
Full table
Furthermore, Ang II significantly induced an increase in α-SMA expression in HK2 cells within 72 h (P<0.01). CTGF-AS could markedly inhibit this effect (P<0.01). In contrast, there were no changes in the scrambled oligonucleotide group (Figure 5, Table 2).
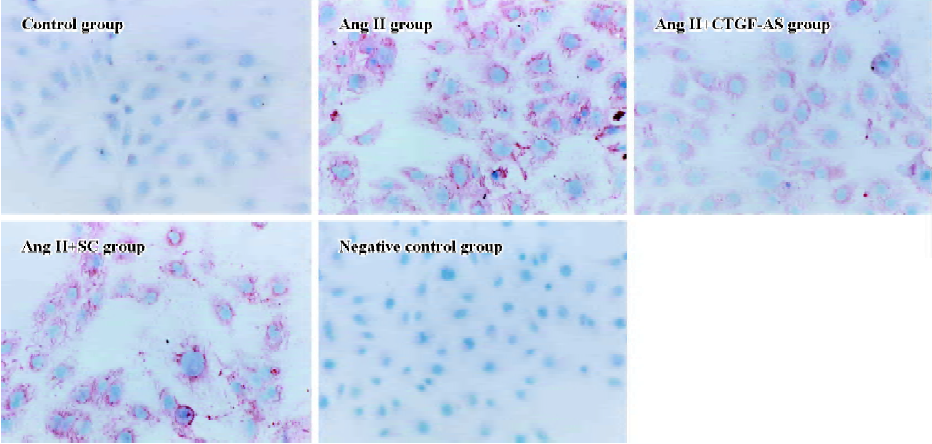
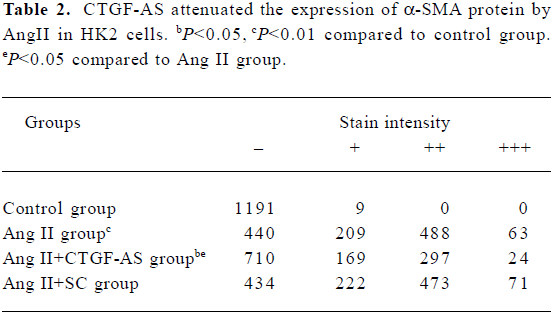
Full table
Discussion
For many years, the importance of tubulointerstitial fibrogenesis has been ignored, and only recently has its important contribution to the onset and progression of renal failure become generally accepted. EMT has been thought to be an important source of myofibroblast in renal interstitium, the latter is regarded as the major contributor of the over-production of ECM[12,13]. The degree of EMT correlated with the severity of tubular damage and subsequent tubulointerstitial fibrosis progression. Ang II plays a central role in mediating the progression of chronic renal interstitial fibrosis. Apart from its pressure effects, Ang II exerts a variety of nonhemodynamic effects that are linked to the renal pathology, which includes induced cell proliferation with subsequent hypertrophy and cell transdifferentiation, and increased the synthesis of the extracellular matrix[4,14].
The current study also demonstrated that AngII could induce the synthesis of fibroblast-specific protein-1 in HK2 cells and the expression of α-SMA. The cellular ultrastructural mesenchymal features were also observed in Ang II-stimulated cells. However, the exact mechanisms of tubular cell transdifferentiation are still unclear. A few studies suggested that numerous growth factors, cytokines, hormones and extracellular cues had been involved in regulating EMT. Strutz et al identified mouse tubular epithelium transfected with FSP-1, a specific fibroblast marker protein, began to show signs of transdifferentiation to a more fibroblast-like phenotype associated with a loss of cytokeratin expression and a gain of vimentin expression[15]. The mouse renal proximal tubule epithelial cells exposed to the cytokines epidermal growth factor and transforming growth factor (TGF)-β had been found transdifferentiated to fibroblast-like cells with increased expressions of collagen, vimentin, FSP-1 and α-SMA[16]. Cultured in 10 or 50 ng/mL TGF-β, Fan et al found that the NRK52E cells presented profound changes. These changes included cellular hypertrophy, loss of apical-basal polarity and microvilli, with cells becoming elongated and invasive, the formation of new front-end back-end polarity, and the appearance of actin microfilaments and dense bodies. These morphological changes were accompanied by phenotypic changes, which involved a loss of the epithelial marker E-cadherin and de novo expression of alpha-SMA[17]. Recently, McMorrow et al demonstrated that cyclosporin A was a direct stimulus for EMT in renal tubule epithelial cells and implicated CTGF as mediators of this response[18]. However, many questions still remain regarding the detailed mechanisms. Ruperez et al found an increased renal CTGF expression with systemic infusion of Ang II into normal rats for 3 d[19]. At d 7, AngII-infused rats presented overexpression of CTGF in glomeruli, tubuli and renal arteries, as well as tubular injury and elevated fibronectin deposition. In addition, treatment with an AT(1) receptor antagonist diminished AngII-induced CTGF and fibronectin overexpression, and ameliorated tubular damage. In cultured renal cells (mesangial and tubular epithelial cells), Ang II, via AT(1) receptor, increased CTGF mRNA and protein production. However, CTGF-AS decreased Ang II-induced fibronectin synthesis, suggesting that CTGF could be a mediator of the profibrogenic effects of Ang II in the kidney. But whether the effect of Ang II on EMT is via CTGF is unknown.
In the current study, we clarified the relationship between Ang II and CTGF in the process of Ang II-induced EMT by using CTGF-AS. Our data demonstrated that Ang II induced the overexpression of CTGF mRNA and protein. In addition, the transdifferential responses induced by Ang II, including the expression of α-SMA and the mesenchymal features in the cellular ultrastructure in HK2 cells, could be significantly prevented by cotreatment with CTGF-AS. Our data strongly suggested that CTGF might be an important mediator of the development of tubular EMT induced by Ang II.
CTGF is a member of the CCN protein family, structurally characterized by their cysteine-rich sequence. Studies have shown that CTGF bears diverse bioactivities, which include triggering mitogenesis, chemotaxis and matrix production. However, CTGF can prevent cell proliferation, stimulate apoptosis or modulate angiogenesis[20]. While CTGF can be found in various human tissues, it is most abundant in the kidney. Increased expression of CTGF has been reported in a variety of glomerular and tubulointerstitial lesions. It has been reported that increased CTGF mRNA expression is found in a variety of fibrotic renal diseases, including IgA nephropathy, crescentic glomerulonephritis, focal glomeru-losclerosis, class IV lupus nephritis, chronic transplant rejection, diabetic glomerulosclerosis and anti-Thy-1.1 nephritis[7,14]. In cultured renal cells, CTGF is upregulated in primary human and rat mesangial cells exposed to high glucose and TGF-β1. Recombinant CTGF induced the expression of extracellular matrix protein[21]. CTGF antisense oligo-deoxynucleotide transfection significantly attenuated TGF-β-induced fibronectin and collagen I mRNA expression[22]. Zhang et al recently reported that rhCTGF could promote the transdifferentiation of human renal tubular epithelial cells towards myofibroblasts in vitro, both directly and as a downstream mediator of TGF-β[23]. Hence, it is generally accepted that CTGF serves as a downstream regulatory factor of TGF-β-induced renal fibrosis. And because of the inflammatory and immunosuppressive properties of TGF-β, CTGF seems to be an attractive alternative therapeutic target against tubulointerstitial fibrosis[24]. Andersen et al[25] found that Losartan, an Ang II-receptor blockade, persistently reduced urinary CTGF excretion, which is associated with a slower rate of decline in GFR in hypertensive type 1 diabetic patients with diabetic nephropathy. van Nieuwenhoven et al showed that a dual blockade of RAS by using ACEI and ARA could more significantly inhibit the urinary excretion of CTGF in type-2 diabetic patients, suggesting that CTGF might be a downstream modulator of Ang II in mediating renal fibrosis[26]. Our results, along with those of other researchers, strongly suggested that CTGF mediated Ang-II induced transdifferentiation of proximal tubular cells and tubulointerstitial fibrosis.
Our data may provide important information that CTGF, as a novel fibrogenic growth factor, mediated Ang-II induced tubular EMT, which is regarded as an important profibrotic factor in progressive tubulointerstitial fibrosis leading to end-stage renal failure. These findings may open up new avenues for therapeutic intervention in the process of tubulointerstitial fibrosis.
References
- Kennefick TM, Anderson S. Role of angiotensin II in diabetic nephropathy. Semin Nephrol 1997;17:441-7.
- Mezzano SA, Ruiz-Ortega M, Egido J. Angiotensin II and renal fibrosis. Hypertension 2001;38:635-8.
- Ruiz-Ortega M, Lorcnzo O, Suzuki Y, Ruperez M, Egido J. Proinflammatory actions of angiotensins. Curr Opin Nephrol Hypertens 2001;10:321-9.
- Wahab NA, Yevdokimova N, Weston BS, Roberts T, Li XJ, Brinkman H, et al. Role of connective tissue growth factor in the pathogenesis of diabetic nephropathy. Biochem J 2001;359:77-87.
- Ito Y, Aten J, Bendc RJ, Ocmar BS, Rabclink TJ, Weening JJ, et al. Expression of connective tissue growth factor in human renal fibrosis. Kidney Int 1998;53:853-61.
- Liu BC, Chen Q, Luo DD, Sun J, Phillips AO, Ruan XZ, et al. Mechanisms of irbesartan in prevention of renal lesion in streptozotocin-induced diabetic rats. Acta Pharmacol Sin 2003;24:67-73.
- Liu BC, Sun J, Chen Q, Ma KL, Ruan XZ, Phillips AO. Role of connective tissue growth factor in mediating hypertrophy of human proximal tubular cells induced by angiotensin II. Am J Nephrol 2003;23:429-37.
- Hannken T, Schroeder R, Stahl RAK, Wolf G. Atrial natriuretic peptide attenuates AngII induced hypertrophy of renal tubular cells. Am J Physiol 2001;281:F81-F90.
- Zeisberg M, Strutz F, Muller GA. Renal fibrosis: an update. Curr Opin Nephrol Hypertens 2001;10:315-20.
- Hishikawa K, Oemar BS, Nakaki T. Static pressure regulates connective tissue growth factor expression in human mesangial cells. J Biol Chem 2001;276:16797-803.
- Ito Y, Goldschmeding R, Bende R, Claessen N, Chand M, Kleij L, et al. Kinetics of connective tissue growth factor expression during experimental proliferative glomerulonephritis. J Am Soc Nephrol 2001;12:472-84.
- Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest 2003;112:1776-84.
- Zeisberg M, Kalluri R. The role of epithelial-to-mesenchymal transition in renal fibrosis. J Mol Med 2004;82:175-81.
- Wolf G, Ziyadeh FN. Molecular mechanisms of diabetic renal hypertrophy. Kidney lnt 1999; 56: 393–405.
- Strutz F, Okada H, Lo CW, Danoff T, Carone RL, Tomaszewski JE, et al. Identification and characterization of a fibroblast marker: Fsp1. J Cell Biol 1995;130:393-405.
- Okada H, Danoff T, Kalluri R, Neilson EG. Early role of FSP1 in epithelial-mesenchymal transformation. Am J Physiol 1997;273:F563-F574.
- Fan JM, Ng YY, Hill PA, Nikolic-Paterson DJ, Mu W, Atkins RC, et al. Transforming growth factor-beta regulates tubular epithelial-myofibroblast transdifferentiation in vitro. Kidney Int 1999;56:1455-67.
- McMorrow T, Gaffney MM, Slattery C, Campbell E, Ryan MP. Cyclosporine A induced epithelial-mesenchymal transition in human renal proximal tubular epithelial cells. Nephrol Dial Transplant 2005;20:2215-25.
- Ruperez M, Ruiz-Ortega M, Esteban V. Angiotensin II increases connective tissue growth factor in the kidney. Am J Pathol 2003;163:1937-47.
- Gupta S, Clarkson MR, Duggan J, Brady HR. Connective tissue growth factor: potential role in glomerulosclerosis and tubulo-interstitial fibrosis. Kidney Int 2000;58:1380-99.
- Riser BL, Denichilo M, Cortes P, Baker C, Grondin JM, Yce J, et al. Regulation of connective tissue growth factor activity in cultured rat mesangial cells and its expression in experimental diabetic glomerulosclerosis. J Am Soc Nephrol 2000;11:25-38.
- Yokoi H, Sugawara A, Mokoyama M, Mort K, Makino H, Suganami T, et al. Role of connective tissue growth factor in profibritic action of transforming growth factor-beta: a potential target for preventing renal fibrosis. Am J Kidney Dis 2001;38:Sl34-S138.
- Zhang C, Meng X, Zhu Z, Liu J, Deng A. Connective tissue growth factor regulates the key events in tubular epithelial to myofibroblast transition in vitro. Cell Biol Int 2004;28:863-73.
- Yokoi H, Mukoyama M, Nagae T. Reduction in connective tissue growth factor by antisense treatment ameliorates renal tubulointerstitial fibrosis. J Am Soc Nephrol 2004;15:1430-40.
- Andersen S, van Nieuwenhoven FA, Tarnow L, Rossing P, Rossing K, Wieten L, et al. Reduction of urinary connective tissue growth factor by losartan in type 1 patients with diabetic nephropathy. Kidney Int 2005;67:2325-9.
- van Nieuwenhoven FA, Rossing K, Andersen S. Beneficial effect of dual blockade of the renin angiotensin system (RAS) on urinary connective tissue growth factor (CTGF) in type 2 diabetic patients with nephropathy. J Am Soc Nephrol 2004;15:571A.