
Ginsenoside Rg1 inhibits proliferation of vascular smooth muscle cells stimulated by tumor necrosis factor-α1
Introduction
Several natural products have been used for vascular diseases and some traditional herbal prescriptions have been employed for treatment of atherosclerosis. Ginsenosides are the most important active constituents identified in ginseng[1], which can protect against atherosclerosis[2]. Ginsenoside Rg1, a steroidal saponin of high abundance in ginseng, can relax the rat aorta and enhance NO production[2–3]. Gene chip analyses find ginsenoside Rg1 can affect the expression levels of genes involved in vascular constriction, cell adherence, coagulation, cell growth and signal transduction in TNF-α stimulated endothelial cells[4]. Atherosclerosis is the principal vascular lesion which is initiated by vascular endothelial cell damage followed by an intimal hyperplasia of VSMC. VSMC change their phenotype from a contractile to a synthetic state and actively proliferate in the intima of atherosclerotic arteries, secreting excess amounts of extra-celluar matrix components including collagen and proteo-glycans[5]. Thus inhibition of VSMC hyperpro-liferation is one of the key pharmacological strategies for prevention of atherosclerosis. TNF-α, a cytokine that is secreted by VSMC in the atherosclerotic neointima, can promote cell growth and movement, both of which are critical for the initiation and progression of vascular lesions[6]. An investigation of ginsenoside Rg1-induced alterations in protein expression in TNF-α-activated VSMC will aid in understanding the molecular mechanism of ginsenoside Rg1.
Proteomics describe all the proteins encoded from a specific genome[7]. Proteomic analysis involves the qualitative alterations in proteins along with the quantitative changes in protein expression levels that occur in response to a given set of conditions[8]. The rapid increase in proteomic-based drug studies stems from the potential benefits associated with the elucidation of drug mechanisms.
As little is known regarding the vascular molecular mechanism of ginsenoside Rg1 on VSMC and because proteomic technique has more advantages in molecular identification, we attempted to use proteomic analysis to explain the molecular mechanism of ginsenoside Rg1 on the proliferation of cultural VSMC.
Materials and methods
Reagents and materials Electrophoresis reagents including acrylamide, methylenebisacrylamide, N,N,N',N',-tetramethylethyl-diamide (TEMED), hydroxymethyl aminomethane (Tris), glycine, sodium dodecyl sulfate (SDS), dithiothreitol (DTT), 3-[3-(cholamidopropyl) dimethyl-ammonio]-1-propane-sulfonate (CHAPS), Immobiline DryStrips, Immobilized pH gradient (IPG) buffer, IPG coverfluid and low molecular marker were purchased from Amersham Biosciences (Sweden). Iodoacetamide and trifluoroacetic acid (TFA) were purchased from Acros (Geel, Belgium). Trypsin was obtained from Boehringer Mannheim (GE, Germany).
TNF-α, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxy-methoxyphenyl)-2-(4-sulfonyl) -2H-tetrazolium (MTS) was obtained from Sigma (St Louis, Mo, USA), ginsenoside Rg1 was obtained from Beijing Institute of Biological Products (Beijing, China), Dulbecco’s modified Eagle’s medium (DMEM) was purchased from Gibco (Los Angeles, USA). Male Sprague-Dawley rats were obtained from the Experimental Animal Center of the Academy of Military Medical Sciences (Beijing, China).
Cell culture Primary VSMC cultures were isolated after removal of the adventitia and scraping of the endothelium. Arterial strips were then incubated in DMEM with 20% fatal bovine serum (FBS), penicillin 100 U/mL, streptomycin 100 mg/L at 37 ºC in 5% CO2 humidified atmosphere. VSMC were propagated in DMEM with 10% FBS in subsequent passages. Mean incubation time was 5 d. Cell viability was confirmed by Trypan blue exclusion after each passage and assay[9]. All assays were conducted with subconfluent cells from passages 3–6.
Proliferation assay VSMC were detached using trypsin and seeded at a density of 5000 cells/well in a 96-well plate and rendered quiescent. Then VSMC were incubated in DMEM with various concentrations of ginsenoside Rg1 and TNF-α for 48 h, and 20 µL of MTS was added. The cultures were incubated for a further 1.5 h, after which the absorbance was read in a multiplate reader. MTS assay is a colorimetric method for determining the number of viable cells in proliferation. It is composed of a solution of tetrazolium, which is bioreduced by metabolically active cells into a soluble formazan product in culture medium. Its absorbance can be measured at 490 nm. The quantity of formazan produced is directly proportional to the number of living cells.
Flow cytometry Aliquots of 5×105 VSMC were centrifuged at 200×g for 5 min, and cell pellets were fixed with 70% ethanol overnight. The cells were then washed twice with PBS and incubated with 1 g/L RNaseA for 30 min. The cells were then resuspended in 0.5 mL of solution containing 50 mg/L propidium iodide and stored in the dark for 20 min. Cells were analyzed on a FACScan Flow Cytometer (FACSCalibur; Becton Dickinson, CA, USA).
Protein extraction and determination of concentrations Following growth arrest, VSMC were incubated in DMEM with various concentrations of ginsenoside Rg1 and TNF-α for 24 h. Cells were then trypsinized and washed with PBS buffer; 1×107 cells were added to 100 µL lysis buffer (8 mol/L urea, 4% CHAPS, 40 mmol/L Tris base). After a few cycles of quick freezing and subsequent thawing in liquid nitrogen, 4 µL RNase (10 g/L) was added. After deposition for 20 min, the cells were centrifuged at 12 000×g for 30 min. The supernatant was transferred and stored. Protein concentrations were determined using the Bradford assay.
Two-dimensional electrophoresis (2-DE) and image analysis Isoelectric focusing (IEF) was carried out on gel strips as described by Ying et al[10]. After IEF separation, the gel strips were immediately equilibrated for two steps in equilibrium buffer, and the second dimension separation was performed using ETTAN DALT six (Amersham Biosciences). The gels were stained and scanned with an ImageScanner (Amersham Biosciences) in transmission mode. Spot detection and matching were performed using ImageMaster 2D Elite 4.01 (Amersham Biosciences). The gel of TNF-α treated cells was selected as a reference gel. Other gels were matched with the reference gel. Values of spot abundance were normalized and exported for statistical analysis.
In-gel digestion and MALDI-TOF-MS identification In-gel digestion of proteins from 2D gels was carried out as described by Steiner et al[11]. Spots were excised and destained with 25 mmol/L ammonium bicarbonate/50% acetonitrile, then dried in a vacuum concentrator (Savant, Holbook, NY, USA). The dried gel pieces were rehydrated with 5 μL of 20 mg/L trypsin solution, and digested at 37 ºC for 18–20 h. Tryptic peptides were extracted using 5% TFA and dried in a vacuum concentrator. MS was performed on a Reflex III MALDI-TOF mass spectrometer (Bruker Daltonics, Bremen, Germany). Database searching was performed in the NCBI database and pI were acquired from image analysis as described by Lim et al[12].
RNA isolation and reverse transcriptase polymerase chain reaction (RT-PCR) Total RNA was extracted from cultured cells according to the supplied protocol. Total RNA was reverse transcripted at 42 ºC for 1 h using reverse transcriptase. The cDNA was amplified using glyseralde-hyde-3-phosphate dehydrogenase(GAPDH) cDNA as an internal control for the amplification of p21 and protein kinase C-ζ (PKC-ζ) expressed by the cells. Primers for the GAPDH were as follows: sense: 5'-ACCACAGTCCATGCC-ATCAC-3'; antisense: 5'-TCCACCACCCTGTTGCTGTA-3'. Primer sequences for p21 were as follows: sense 5'-CCGATA-CAGGTGATGATGATGG-3', antisense: 5'-GCCAGAAGTG-AAGCCAAGG-3'; Primer sequences for PKC-ζ were as follows: sense: 5'-GCCTCCAGTAGATGACAAGAAC-3', antisense: 5'-GAGTGTAAGCCAACCAGGAAG-3'. The sample was amplified for 28 cycles with the following parameters: 94 ºC 30 s, 58 ºC 20 s, 72 ºC 30 s. The PCR products were subjected to 2% agarose gel electro-phoresis. Quantitative data normalized to GAPDH were obtained from a densitometer and analyzed with the included Quantity One 4.4.0 software (BIO-RAD, USA).
Statistical analysis Data was reported as mean±SD. One-way ANOVA or Student's t-test was used for statistical analysis of the original data, and significance was accepted at P<0.05.
Results
Effects of ginsenoside Rg1 on proliferation of TNF-α stimulated VSMC Following growth arrest, VSMC were stimulated with TNF-α for 24 h. TNF-α induced VSMC proliferation significantly compared with the control. However, ginsenoside Rg1 inhibited VSMC growth at doses as low as 20 mg/L (Figure 1). This observation was not a result of cell necrosis as ginsenoside Rg1-treated cells remained greater than 90% viable after Trypan blue staining. The effects of ginsenoside Rg1 on cell cycle progresssion were determined by flow cytometry (Table 1). Quiescent VSMC was induced to enter S phase by stimulation with TNF-α. The population of G0/G1 cells decreased with a significant concomitant rise in S phase cells. Ginsenoside Rg1 significantly inhibited VSMC from G1 to S progresssion, as shown by the increase in G0/G1 cells accompanied by a concurrent decrease in S phase cells. Therefore, ginsenoside Rg1 not only inhibited VSMC proliferation, but also arrested VSMC in G0/G1.
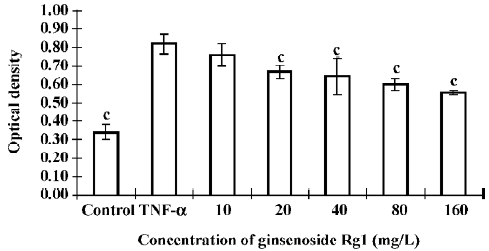
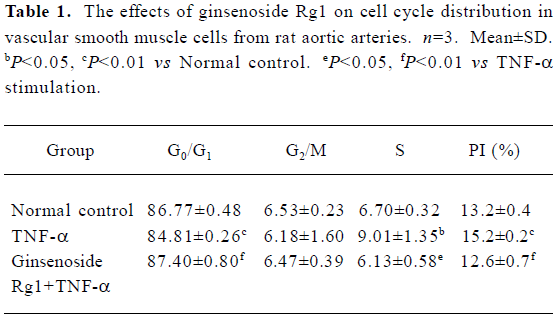
Full table
Protein expression in normal, TNF-α-stimulated and ginsenoside Rg1-treated VSMC Protein samples extracted from normal, TNF-α-stimulated and ginsenoside Rg1-treated VSMC were loaded on IPGphor gel strips for IEF. Vertical SDS-PAGE was performed immediately following IEF. This experiment was repeated three times. A representative batch of gels is shown in Figure 2. Two dimentional gels of normal, TNF-α-stimulated and ginsenoside Rg1-treated VSMC were analyzed for the purpose of quantitative spot comparisons. Analyzed results showed 845±22 spots in a normal cell map, 892±242 spots in a TNF-α-stimulated cell map, and 879±21 spots in a ginsenoside Rg1-treated cell map. The gels of TNF-α stimulated cells were selected as reference gels, the gels of normal and ginsenoside Rg1 treated cells were matched to reference gels. Differential analysis showed 24 protein spots were different in intensity, including 17 spots that were increased and 7 spots that were decreased after TNF-α stimulation. Ginsenoside Rg1 treatment could prevent this change or reverse it to some degree (Figure 2, Table 2).
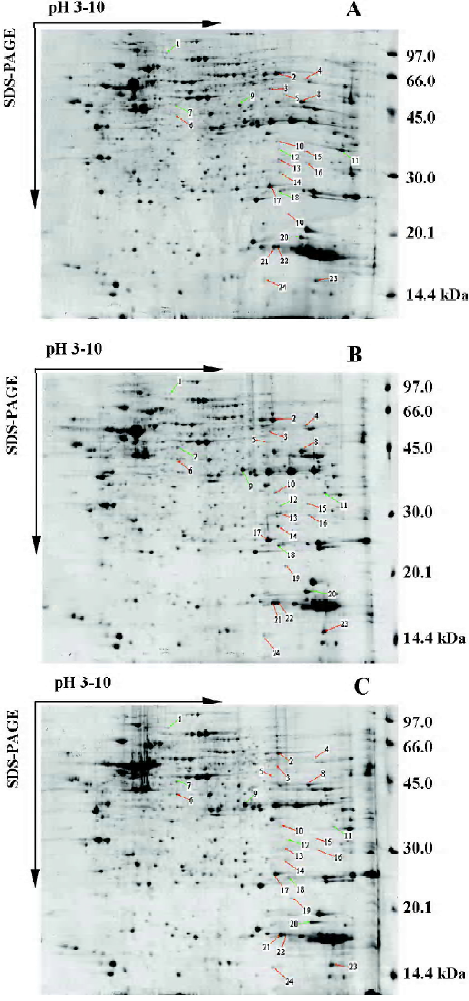
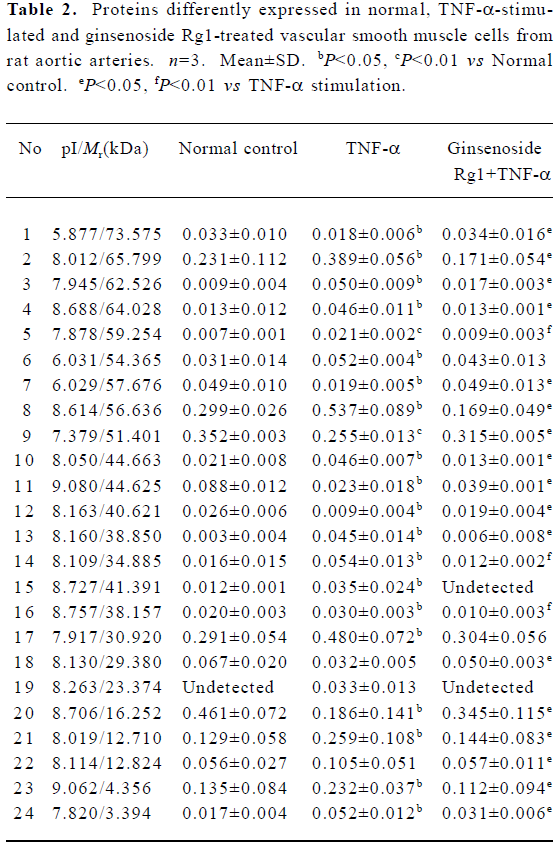
Full table
Identification of proteins associated with different expressions in normal and ginsenoside Rg1-treated VSMC compared with TNF-α-stimulated VSMC by PMF The differently expressed spots were excised from stained Coomassie Brilliant R-250 gels and submitted to in-gel tryptic digestion. The resulting peptide mixtures were analyzed by MALDI-TOF-MS. The resulting peptide mass maps were used to search against a comprehensive protein sequence database, typical peptide mass maps were listed in Figure 3. Table 3 lists the proteins expressed differently in normal and ginsenoside Rg1-treated VSMC compared with TNF-α-stimulated VSMC. We identified 8 proteins by database searching according to identified standards[12]. Other proteins need to be investigated further because of their poor score. The expression of G-protein coupled receptor kinase, PKC-ζ, N-ras protein were increased, while cycle related protein p21, was decreased by TNF-α compared with normal VSMC. Ginsenoside Rg1 could restore the expression levels of these proteins, at least in part, to basic levels of untreated cells.
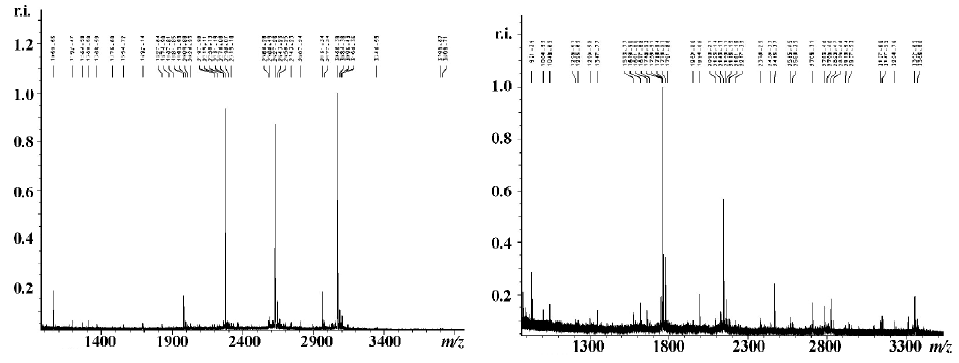
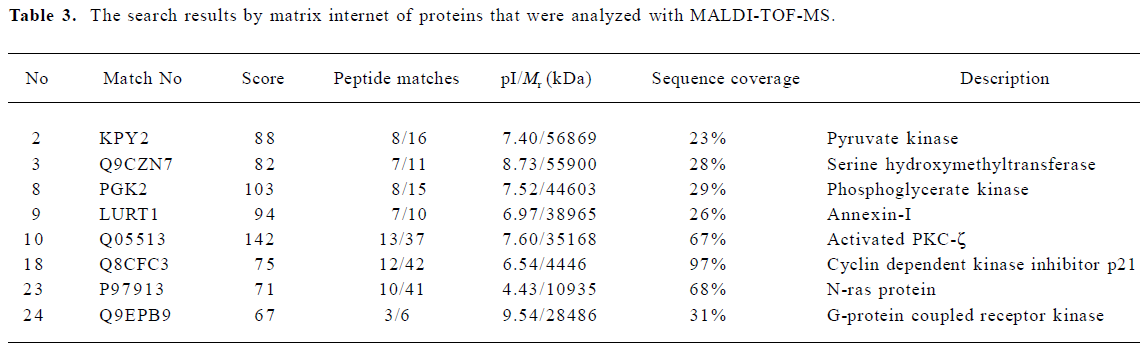
Full table
Effects of ginsenoside Rg1 on mRNA expression in VSMC TNF-α down-regulated more than 60% of p21 mRNA expression compared with normal VSMC, while ginsenoside Rg1 upregulated more than 50% of p21 mRNA expression compared with TNF-α stimulated VSMC. The effects of ginsenoside Rg1 on PKC-ζ mRNA expression in VSMC had contrary changes. TNF-α upregulated PKC-ζ to more than 40% of mRNA expression compared with normal VSMC, while ginsenoside Rg1 down-regulated PKC-ζ to more than 17% of mRNA expression compared with TNF-α stimulated VSMC (Figure 4).
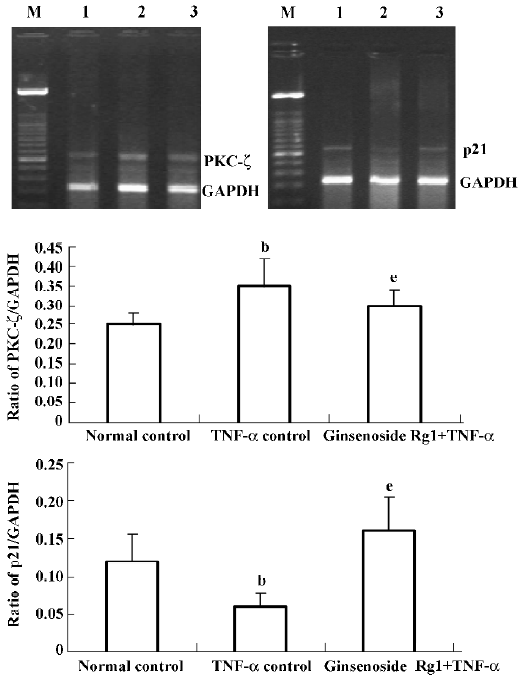
Discussion
VSMC in healthy vessel walls have low mitogenic activity. During the early stages of arterial wall injury or athero-sclerosis, VSMC may undergo a phenotypic transformation[13]. They become activated and then proliferate and migrate to the intimal layer where they accumulate lipids and participate in plaque formation atherosclerosis[14]. The inhibition of this process is considered to be of great benefit in the maintenance of vascular homeostasis and in the prevention and development of atherosclerosis. Both ischemic and direct vascular injury result in the elaboration of proinflam-matory substances, including TNF-α, which regulate VSMC proliferation and promote vessel stenosis[15]. Other investigators have suggested that TNF-α induces VSMC proliferation[16]. In our present study, we found that TNF-α stimulated VSMC proliferation. Ginsenoside Rg1 is a major ingredient used in many traditional Chinese medicines to cure atherosclerosis. It has biphasic regulation and little adverse effect. Our results suggest that ginsenoside Rg1 inhibits the proliferation of VSMC in a dose-dependent manner and this action is not due to a cell-toxicity effect.
There have been no previous studies to report that ginsenoside Rg1 has the action to inhibit proliferation of VSMC. Cell-cycle analysis using flow cytometry confirmed the results of MTS methods. Flow cytometry results showed that ginsenoside Rg1 arrested the VSMC cell cycle at G1 in growing-phase cells and inhibited the cell cycle progression from G0/G1 to S phase in G0-arrested cells. These results suggest that the inhibitory effect of ginsenoside Rg1 is exerted at a point in the G1 phase. It has been reported that cyclin dependent kinase (CDK) promote G1/S transition and that p21 is an intrinsic inhibitor of the kinase activity of CDK[17]. At 40 mg/L, ginsenoside Rg1 increased the p21 protein level while producing G1 arrest. RT-PCR results suggested ginsenoside Rg1 increased p21 mRNA levels. These results indicate that the induction of p21 by ginsenoside Rg1 is due to an induced increase in its mRNA level. It has recently been reported that the migration of VSMC can be inhibited by an overproduction of p21 in cultured VSMC[18], thus the induction of p21 by ginsenoside Rg1 may be one of the mechanisms by which ginsenoside Rg1 exerts its action on migration activity.
PKC could mediate activation of extracellular signal-regulated kinase (ERK) and mitogen-activated protein kinase (MAPK) in VSMC. The proliferation of VSMC is mediated by the MAPK pathway[19]. The down-regulation of PKC-ζ by ginsenoside Rg1 may be one of the mechanisms that inhibit proliferation. PKC-ζ, an atypical PKC that are Ca2+-independent and do not require diacylglycerol for activation, can phosphorylate and decrease the half-life of p21[20]. PKC also play a role in p21 mRNA stability[21], which hints that activation of PKC-ζ results in a decrease of the p21 level. The induction of p21 and inhibition of PKC-ζ may be the mechanism by which ginsenoside Rg1 inhibits VSMC proliferation.
Because upregulation of N-ras activated MAPK pathway and induced inflammation[22–23], ginsenoside Rg1 could downregulate the N-ras level, which suggests it exerts anti-inflammation and anti-proliferation effects by the N-ras pathway.
The present results suggest that ginsenoside Rg1 inhibits the proliferation of VSMC through an induction of p21 and inhibition of PKC-ζ. The above results suggest that ginsenoside Rg1 is proved to be a valuable drug to cure atherosclerosis.
References
- Attele AS, Wu JA, Yuan CS. Ginseng pharmacology: multiple constituents and multiple actions. Biochem Pharmacol 1999;58:1685-93.
- Chen X. Cardiovascular protection by ginsenosides and their nitric oxide releasing action. Clin Exp Pharmacol Physiol 1996;23:728-32.
- Kang SY, Schini-Kerth VB, Kim ND. Ginsenosides of the protopanaxatriol group cause endothelium-dependent relaxation in the rat aorta. Life Sci 1995;56:1577-86.
- Lv JP, Ma ZC, Yang J, Huang J, Wang SR, Wang SQ. Ginsenoside Rg1-induced alterations in gene expression in TNF-α stimulated endothelial cells. Chin Med J 2004;117:871-7.
- Kaji T, Okabe M, Shimada S, Yamamoto C, Fujiwara Y, Lee JB, et al. Sodium spirulan as a potent inhibitor of arterial smooth muscle cell proliferation in vitro. Life Sci 2004;74:2431-9.
- Faries PL, Rohan DI, Wyers MC, Marin ML, Hollier LH, Quist WC, et al. Vascular smooth muscle cells derived from atherosclerotic human arteries exhibit greater adhesion, migration and proliferation than venous cells. J Surg Res 2002;104:22-8.
- Wilkins MR, Sanchez JC, Gooley AA, Appel RD. Progress with proteome projects: why all proteins expressed by a genome should be identified and how to do it. Biotechnol Genet Eng Rev 1996;13:19-50.
- Anderson NL, Anderson NG. Proteome and proteomics: new technologies, new concepts, and new words. Electrophoresis 1998;19:1853-61.
- Yong ZH, Jie Y, Guang RZ, Ying JY. Ferulic acid inhibits vascular smooth muscle cell proliferation induced by angiotensin II. Eur J Pharmacol 2004;499:85-90.
- Ying W, Zhang K, Qian X, Xie L, Wang J, Xiang X, et al. Proteome analysis on an early transformed human bronchial epithelial cell line, BEP2D, after alpha-particle irradiation. Proteomics 2003;3:64-72.
- Steiner S, Gatlin CL, Lennon JJ, McGrath AM, Aponte AM, Makusky AJ, et al. Proteomics to display lovastatin-induced protein and pathway regulation in rat liver. Electrophoresis 2000;21:2129-37.
- Lim H, Eng J, Yates JR, Tollaksen SL, Giometti CS, Holden JF, et al. Identification of 2D-gel proteins: a comparison of MALDI/TOF peptide mass mapping to mLC-ESI tandem mass spectrometry. J Am Soc Mass Spectr 2003;14:957-70.
- Rivard A, Andres V. Vascular smooth muscle cell proliferation in the pathogenesis of atherosclerotic cardiovascular diseases. Histol Histopathol 2000;15:557-71.
- Ferguson JE, Patterson C. Break the cycle: the role of cell-cycle modulation in the prevention of vasculoproliferative diseases. Cell Cycle 2003;2:211-9.
- Boehm M, Nabel EG. The cell cycle and cardiovascular diseases. Prog Cell Cycle Res 2003;5:19-30.
- Lee B, Moon SK. Resveratrol inhibits TNF-alpha-induced proliferation and matrix metalloproteinase expression in human vascular smooth muscle cells. J Nutr 2005;135:2767-73.
- Yu CH, Wu J, Su YF, Ho PY, Liang YH, Sheu MT. Anti-proliferation effect of 3-amino-2-imino-3,4-dihydro-2H-1,3-benzothiazin-4-one (BJ-601) on human vascular endothelial cells: G0/G1 p21-associated cell cycle arrest. Biochem Pharmacol 2004;67:1907-16.
- Fukui R, Shibata N, Kohbayashi E, Amakawa M, Furutama D, Hoshiga M, et al. Inhibition of smooth muscle migration by the p21 cyclin-dependent kinase. Atherosclerosis 1997;132:53-9.
- Ginnan R, Pfleiderer PJ, Pumiglia K, Singer HA. PKC-delta and CaMKII-delta mediate ATP-dependent activation of ERK1/2 in vascular smooth muscle. Am J Physiol Cell Physiol 2004;286:C1281-9.
- Scott MT, Ingram A, Ball KL. PDK1-dependent activation of atypical PKC leads to degradation of the p21 tumour modifier protein. EMBO J 2002;21:6771-80.
- Park JW, Jang MA, Lee YH, Passaniti A, Kwon TK. p53-Independent elevation of p21 expression by PMA results from PKC-mediated mRNA stabilization. Biochem Biophys Res Commun 2001;280:244-8.
- Yang CM, Chien CS, Hsiao LD. Mitogenic effect of oxidized low-density lipoprotein on vascular smooth muscle cells mediated by activation of Ras/Raf/MEK/MAPK pathway. Br J Pharmacol 2001;132:1531-41.
- Minamino T, Yoshida T, Tateno K, Miyauchi H, Zou Y, Toko H, et al. Ras induces vascular smooth muscle cell senescence and inflammation in human atherosclerosis. Circulation 2003;108:2264-9.