
TRPC3 is involved in flow- and bradykinin-induced vasodilation in rat small mesenteric arteries1
Introduction
Bradykinin locally synthesized in the cardiovascular tissue and blood plasma is believed to contribute to the regulation of cardiovascular homeostasis such as vascular tone control and vascular permeability alteration by stimulating the endothelial cells to release vasodilators including nitric oxide (NO), prostacyclin (PGI2) and endothelium-derived hyperpolarizing factors. In endothelial cells, it is generally considered that the binding of bradykinin to its receptors – B1 and/or B2 receptors, which are G-protein coupled receptors, activates phospholipase C to catalyze the hydrolysis of PIP2 to yield IP3 and DAG. A biphasic elevation of endothelial [Ca2+]i is observed upon bradykinin stimulation; a transient initial peak followed by a sustained plateau phase. The initial transient rise is contributed by the Ca2+ release from the internal stores via the interaction of IP3 with IP3 receptor in sarcoplasmic/endoplasmic reticulum, whereas the sustained phase is contributed by the extracellular Ca2+ influx through Ca2+ permeable channels in the plasma membrane[1–6].
Flow shear stress is the most important physiological stimulus for the synthesis and release of different potent vasodilators from endothelial cells to induce vasodilation. Flow may elicit vasodilation either via a [Ca2+]i-dependent mechanism[3,5] or a [Ca2+]i -independent mechanism[2]. Flow-induced Ca2+ influx may be attributed to activation of mechanosensitive cation channels[27], stimulation of P2X4 purinoceptor [24,25], modulation of cytoskeleton-mediated signal transduction pathways[2], activation of K channels, causing an increased driving force for extracellular Ca2+ influx[20], or facilitation of the delivery of agonists (eg, ATP, ADP and bradykinin) to the unstirred boundary layer at the cell surface[4].
Until now, the Ca2+-permeable channel(s) involved in bradykinin- and flow-induced Ca2+ influx in vascular endothelial cells has still not been very clear, but the members of transient receptor potential (TRP) superfamily offer likely candidates[26]. TRP was first described as a Drosophila mutant that had an impaired visual transduction with transient receptor potential in response to continuous light. Since the cloning of Drosophila Trp gene in 1989, approximately 30 unique members of TRP superfamily have been identified on the basis of amino acid sequence and structural similarity. They are classified into TRPC (C for canonical), TRPV (V for vanilloid), TRPM (M for melastatin), TRPP (P for PKD), TRPML (ML for mucolipin) and TRPN (N for NOMPC) subfamilies[10]. All TRP channels, except TRPM4 and TRPM5, are cation channels that allow Ca2+ influx[26]. The TRPC subfamily is composed of seven mammalian members designated to TRPC1 to 7. All these seven TRPC channels are involved in Ca2+ influx in response to activation of PLC by membrane receptors[10].
Targeted knockout of TRPC4 has been shown to markedly reduce the agonist (ATP, acetylcholine and thrombin)-induced Ca2+ influx in mice artery endothelial cells[7,23]. Endothelium-dependent smooth muscle relaxation in response to vasoactive agonists ATP and acetylcholine is also impaired in these mice[7]. These data provide compelling evidence for the key role of TRPC4 in agonist-induced endothelial Ca2+ influx and vascular relaxation. Besides TRPC4, several other TRP channels including TRPC1 and TRPC3 have been suggested to be involved in the agonist-induced Ca2+ influx in endothelial cells[12,13]. For TRPC3, it has been shown that bovine pulmonary artery endothelial cells transfected with TRPC3 channels display increased Ca2+ influx in response to ATP and bradykinin[12]. In addition to agonist-induced Ca2+ influx, TRP channels may also participate in flow-induced Ca2+ influx. For example, shear stress activates TRPV4 in TRPV4-expressing HEK cells[8] and it also activates the complex of TRPP1 and TRPP2 in renal epithelial cells[8,19]. Unfortunately, there is still no evidence to show the role of TRPV4 or TRPP1/TRPP2 complex in flow-induced Ca2+ influx in vascular endothelial cells. More importantly, the role of most TRP channels in agonist- or flow-induced dilatation has not been established, except for the case of TRPC4 and TRPV1, which are known to participate in the vasodilation in response to ATP, acetylcholine and ananda-mine[7,26].
In the present study, we explored the functional role of TRPC3 channel in flow- and agonist-induced dilatation of isolated rat small mesenteric arteries using a specially designed antisense oligo against TRPC3. We also examined the role of TRPC3 in bradykinin- and flow-induced Ca2+ influx in the endothelial cells of isolated small mesenteric arteries.
Materials and methods
Animals Male Sprague-Dawley rats were supplied by the Laboratory Animal Service Centre of the Chinese University of Hong Kong, Hong Kong, China. We followed the Guide for the Care and Use of Laboratory Animals published by the US Institute for Laboratory Animal Research, National Research Council in 1996.
Preparation of rat small mesenteric arteries Male Sprague-Dawley rats (~250–280 g) were killed by inhalation of CO2. Ileum and associated mesentery were removed and immersed in Krebs-Henseleit solution gassed with 95% O2–5% CO2 mixture. A third- or fourth-order mesenteric artery (~2–3 mm long) was carefully dissected free of surrounding adipose tissue under a dissecting microscope (Nikon, Japan).
Isobaric diameter experiments-measurement of flow-induced vasodilation The method for the flow experiments was as described in a previous study[16]. After dissecting, the mesenteric artery (~3-mm long) was mounted on a pressure myograph (Danish MyoTechnology, Denmark) with two glass micropipettes and both cannulation pipettes were connected to independent reservoirs set at the same height and solution level to ensure no flow. Changes in the vessel diameter and pressure were tracked and measured with MyoView software (version 1.1 P, 2000, Photonics Engineer-ing). After being cannulated onto micropipettes, the isolated artery was left to stabilize in Tyrode’s solution while the Tyrode’s solution was continuously superfused around the artery with a peristaltic pump at a rate of 2–3 mL/min and bubbled with pure O2 for at least 30 min before experiments began. BSA 1% was included in intraluminal solution and ATP (1 mmol/L) was routinely added in both the extraluminal and intraluminal solutions. Temperature was kept at 37 °C (± 0.5 °C) in flow experiments.
After the arterial viability was assessed by contraction with 3 mmol/L phenylephrine and subsequent dilation to 1 mmol/L acetylcholine, the viable artery was continued to flow procedure after washing and incubation. The flow (shear stress 3.5–10.7 dynes/cm2) was initiated by a pressure gradient (5–6 mmHg) in phenylephrine-preconstricted artery, by moving the two reservoirs an equal distance (5 cm) but in opposite directions at the same time. This ensured that the change in flow did not cause a simultaneous change in the transmural pressure. The flow was then stopped by moving the two reservoirs back to the same level after observation. The mean intraluminal pressure was maintained at 50 mmHg throughout the flow protocol.
Isometric tension experiments-measurement of agonist-induced vasodilation An approximately 2 mm long segment of mesenteric artery was dissected and transferred to chambers of a Multi Myograph System (Danish MyoTechnology) filled with 95% O2–5% CO2 mixture bubbled Krebs-Henseleit solution. The artery segments were mounted in Mulvany-Halpern myograph (model 400A or 610M, Danish Myo-Technology) with gold-plated tungsten wire (25 µm diameter, Goodfellow, England). Each wire was fixed to mounting jaws of the myograph. Changes in isometric force were continuously recorded by using Maclab software (version 3.5). The chamber solution was continuously gassed with a 95% O2–5% CO2 mixture at 37 °C (pH 7.4). All arteries were set to an optimal resting tension of 1.5 mN, which had been determined by length–tension relationship experiments.
Before commencement of the experiments, all artery segments were allowed to equilibrate for 60 min during which the bath solution was replaced every 20 min with pre-warmed and gassed Krebs-Henseleit solution. The resting tension was readjusted to 1.5 mN when necessary.
After equilibration, endothelial cell viability was assessed as sustained maximal relaxation (>95%) to 1 µmol/L acetylcholine in arteries constricted with submaximal concentrations of phenylephrine (0.5 µmol/L) combined with U46619 (0.05 µmol/L). The healthy segments were then washed with pre-warmed Krebs-Henseleit solution several times (>4 times) until baseline tone restored. Subsequently, the accumulated concentration-response curves of histamine and bradykinin were then evaluated in preconstricted arteries with phenylephrine (0.5 µmol/L) plus U46619 (0.05 µmol/L). For ATP and CPA, we examined the non-accumulated concentration effect, meaning that a single concentration was used. There was a 10–15 min wait before washing and the next concentration was applied.
Measurement of endothelial [Ca2+]i-MetaFluor imaging system In separate experiments, a specifically modified flow chamber from pressure myograph chamber (Danish MyoTechnology) by the Technical Services Unit, Chinese University of Hong Kong was used, which ensured the chamber fit to the MetaFluor imaging system. Following equilibration of pressurized arteries in the flow chamber, 200 µL Tyrode’s solution containing the fluorescent indicator Fluo-4 AM (Molecular Probes, 20 µmol/L) and 0.02% pluronic F127 was pumped into the artery lumen at a pressure of <10 mmHg and kept in the lumen for 1 h in the dark at room temperature. The pressure was kept low during the dye loading to ensure the dye could be selectively loaded into endothelial cells. After the dye loading, the pressure was raised to 50 mmHg and the artery was allowed to stabilize for another 10 min. The chamber was placed on an inverted microscope (IX70, Olympus, NY, USA), equipped with a 20X Olympus water immersion objective (0.50W, UMPLANFL, Olympus). Fluorescence intensity was measured with a MetaFluor imaging system (Universal Imaging, West Chester, PA, USA). The Fluo-4 AM loaded artery was excited at 490 nm and the images of the respective 510 nm emissions were collected at every 2-s interval using MetaFluor v4.6 software (Universal Imaging). The emitted light was transmitted to the collecting device and then to a cooled charge coupled device (CCD) camera (Photometrics Quantix, Roper Scientific, Trenton, NJ, USA). Video frames containing fluorescence images were digitized at 512×480 pixel resolution and then analyzed with MetaFluor.
Antisense oligo treatment The second generation of antisense oligo modified by phosphothioates (s) at both ends of the sequence for three nucleotides was used in the present study. An antisense oligo (antisense-TRPC3) specially targeting the region surrounding the AUG starting codon for rat TRPC3 mRNA was designed by us and synthesized by Tech Dragon, Hong Kong, China. Its sequence was as follows: 5'-gsCsAsTCTAgTTAAsAsgsC-3'. A reversed sequence of the antisense oligo was used as a control: 5'-CsgsAsAATTgATCTsAsCsg-3'.
Antisense oligos or control oligos at 100 μg were directly delivered into the rat blood stream by tail vein injection. After 72-h treatment, the rats were killed and their mesenteric arteries were collected. Then flow- and agonist-induced vasodilations were separately evaluated using a pressure myograph or a multi myograph system. In addition, a few arteries were prepared for immunohistochemical staining for assessment of the expression level of TRPC3.
Immunohistochemical staining of TRPC channels After excision, the small mesenteric arteries were fixed in 10% neutral buffered formalin solution (Sigma, St Louis, USA) overnight, and then embedded with paraffin following a series of dehydration. Tissue sections (4-µm thick) were used in this study. After deparaffination and rehydration, tissue sections were stained with appropriate anti-TRPC polyclonal antibody using a standard SABC method. Tissue sections were permeabilized with PBST and incubated with primary antibody overnight at 4 °C. Secondary antibody [biotinyl-ated-goat anti-rabbit IgG (H+L)] was applied for 1 h at room temperature. After washing with PBS 3 times, HRP-streptavidin conjugate was added onto tissue slides and 1-h incubation was allowed at room temperature. The color was then developed using DAB-H2O2 solution (0.3 g/mL DAB in PBS with 0.003% H2O2) and the cell nuclei were subsequently counter-stained with Mayer’s haematoxylin. Slides were viewed using a Leica microscope (Wetzlar, Germany).
Two negative controls were set for each tissue section. One was the competitive control that was used to test the specificity of the antibody and the other was the blank control that was applied to monitor the whole staining process. The competitive control was prepared by incubating the antibody with its respective antigen (provided by Alomone with antibody) in 1:1 weight ratio for 2.5 h at room temperature and the supernatant was collected after centrifugation at 13 200 rpm for 5 min, proceeding to the same procedure in parallel with primary antibody staining. The blank control was designed by replacing the primary antibody with PBS while staining.
SDS/PAGE and immunoblots Immunoblots were performed as described in a previous study[14]. Rat aortas were homogenized and the lysates were extracted with protein extraction buffer, which contained 50 mmol/L Tris-HCl, 150 mmol/L NaCl, 1% Nonidet P-40, 0.1% SDS, 50 mmol/L NaF, 2 mmol/L EDTA, 0.5% sodium deoxycholate, pH 7.5, with the addition of complete protease inhibitor cocktail tablets. Protein concentrations were determined by Bradford assay (Bio-Rad). Proteins 100 µg were loaded onto each lane and separated on an 8% SDS/PAGE gel after being boiled in SDS loading buffer. After electrophoresis, proteins were transferred to a PVDF membrane, and the membrane was then immersed in a blocking solution containing 5% non-fat milk and 0.1% Tween 20 in PBS buffer for 1 h at room temperature with constant shaking. The incubation with the primary anti-TRPC3 antibodies (1:200 dilution) was carried out overnight in PBS buffer containing 5% non-fat milk and 0.1% Tween 20. Immunodetection was accomplished with horseradish peroxidase (HRP)-conjugated secondary antibody, followed by ECL Plus Western Blotting Detection system. The intensity of the bands was analyzed by the FluorChem 8000 imaging system.
Chemicals and solutions Anti-TRPC antibodies were purchased from Alomone Laboratories (Jerusalem, Israel). Phenylephrine hydrochloride was purchased from RBI. U46619 was obtained from Tocris (Mo, USA). Bradykinin was from Calbiochem (San Diego, USA). Fluo-4 AM (acetoxymethyl ester) and Pluronic F127 were from Molecular Probes . Other chemicals were from Sigma.
Krebs-Henseleit solution contained: NaCl 119 mmol/L, NaHCO3 25 mmol/L, MgCl2 1 mmol/L, KCl 4.7 mmol/L, CaCl2 2.5 mmol/L, KH2PO4 1.2 mmol/L, and D-glucose 11 mmol/L. Tyrode’s solution contained: NaCl 117 mmol/L, MgCl2 1 mmol/L, KCl 4.7 mmol/L, KH2PO4 1.2 mmol/L, CaCl2 1.6 mmol/L, HEPES 10 mmol/L, D-mannitol 30 mmol/L, and D-glucose 11 mmol/L, pH 7.4. PBS contained: NaCl 140 mmol/L, Na2HPO4 10 mmol/L, KCl 3 mmol/L, and KH2PO4 2 mmol/L, pH 7.4. PBST contained 0.01% Tween-20 in PBS, pH 7.4.
Data analysis Vasodilation to flow was calculated as a percentage using the following equation:
% vasodilation=100×(Df–Dphe) / (Di–Dphe),
where D represents vessel external diameter; Df is the maximum vessel diameter during flow; Dphe is the diameter after phenylephrine constriction and before flow; Di is the initial diameter before phenylephrine constriction without any treatments.
For measurement of agonist-induced relaxation, EC50 values were calculated as the agonist concentration that caused 50% maximum relaxation. The effects of agonists were expressed as percentage relaxation from phenylephrine plus U46619-induced contraction. Concentration-relaxation relationship was analyzed with a non-linear regression curve fitting (GraphPad Prism, version 3.0, San Diego, CA).
For Ca2+ imaging, fluorescence intensity before the initiation of flow or the application of bradykinin was normalized to 1 (F0). The responses to flow or bradykinin were displayed as the ratio of fluorescence relative to the intensity before flow (F1/F0).
Statistical evaluation of the effect of antisense oligos was made using a two-tailed Mann-Whitney test by comparing the maximal % of vasodilation to flow, EC50 for agonist-induced relaxation, and F1/F0 for endothelial [Ca2+]i measurement. All data were shown as means±SEM of n experiments on the vessel segments prepared from different rats. Significance was assumed at P<0.05.
Results
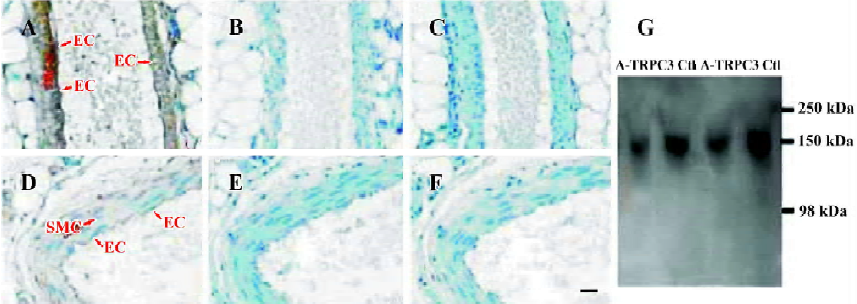
Effect of antisense oligo on TRPC3 expression In order to study the possible involvement of TRPC3 in flow- and agonist-induced vasodilation, TRPC3 antisense oligo (antisense-TRPC3) or its control oligo was injected into the rat tail vein. After 72-h treatment, the inhibitory efficiency of antisense-TRPC3 was evaluated using immunohistochemical staining. Immunohistochemical staining showed that TRPC3 proteins were expressed in both endothelial cells and smooth muscle cells in small mesenteric arteries of rats that were treated with control oligos (100 μg) for 72 h (Figure 1A). In contrast, in rats treated with the antisense-TRPC3, no positive signal was observed in endothelial cells and only weak signals were found in smooth muscle cells (Figure 1D). Two additional control experiments were performed to verify the specificity of immunohistochemical staining, one with antigen preabsorption (Figure 1B,1E) and the other in the absence of primary anti-TRPC3 antibody (Figure 1C,1F). No signals were observed in both sets of controls (Figure 1B,1C,1E,1F). These results suggested that the injected antisense-TRPC3 was effective in suppressing TRPC3 protein expression in endothelial cells as well as smooth muscle cells.
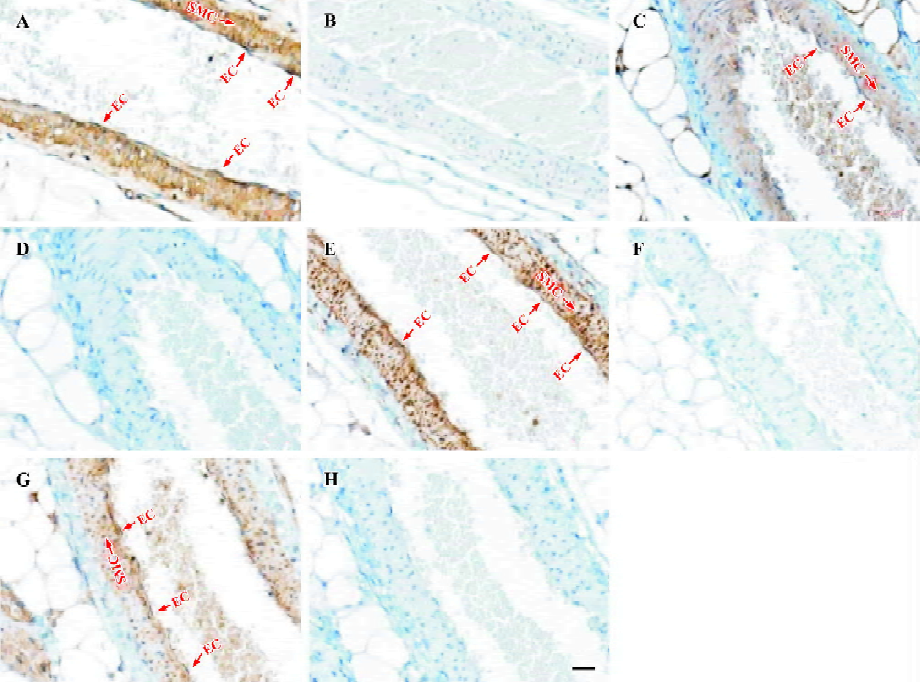
Cellular localization of other TRPC homologs in vascular tissues was explored with immunohistochemical staining using antibodies against other TRPC homologs in small mesenteric arteries. Strong positive signals in brown color could be also observed in vascular endothelial cells as well as in vascular smooth muscle layer for TRPC1, 4–6 homologs (Figure 2A,2C,2E,2G). No signal was observed in competitive controls (Figure 2B,2D,2F,2H) and blank controls (data not shown). The differences in all TRPC staining between positives and competitive controls were very large and obvious, indicating that the immunohistochemical stainings were TRPC-specific. Note that the blue color in experiments and in controls was due to hematoxylin counterstaining, which stained the cell nucleus.
Immunoblots were performed to verify the suppressing effect of antisense-TRPC3 on the expression level of TRPC3 proteins. Because of the difficulty in isolating enough proteins from small mesenteric arteries, proteins from rat aortas were used for the purpose. Immunoblots detected a single band with the molecular size of approximately 150 kDa, which may represent a glycosylated form of TRPC3. Several previous publications have also reported TRPC3 proteins of similar molecular sizes[15,22]. Figure 1G shows that antisense-TRPC3 markedly suppressed the expression of TRPC3 proteins in rat aorta with protein levels decreased by 20%. Note that the aorta contains many layers of vascular smooth muscle cells, which are not in direct contact with circulating blood, therefore the suppressing effect of antisense TRPC3 on the expression of TRPC3 in the aorta is expected to be smaller than that in small mesenteric arteries.
Effect of antisense oligo on flow-induced vasodilation Flow-induced vasodilation was measured using a pressure myograph system. Small mesenteric arteries treated with antisense-TRPC3 or control oligos pressurized to 50 mmHg had an external diameter of 350–400 µm (n=45). After preconstriction with phenylephrine (0.5–1.5 µmol/L) to a similar level (65%–75% of its initial diameter), the artery was exposed to an intraluminal flow initiated by a pressure gradient (5–6 mmHg) to evoke dilation. Flow dilation consisted of an initial transient peak followed by a sustained plateau phase (>10 min), which was flow-dependent. The vessels rapidly contracted again as soon as the flow stopped (Figure 3A). In this study, the peak dilation in response to flow was compared between the control group and the antisense-TRPC3 group. Analysis revealed that, compared to the control oligo treatment, antisense-TRPC3 treatment significantly inhibited the magnitude of flow-induced vasodilation in rat small mesenteric arteries. The maximal percentage of dilation was reduced from the control value of 85.8%±4.5% to 73.1%±2.7%, a reduction of approximately 13% (Figure 3B). These results suggested an involvement of TRPC3 in flow-induced vasodilation.

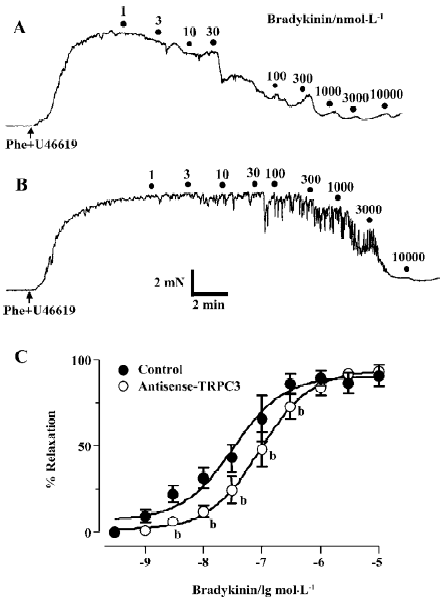
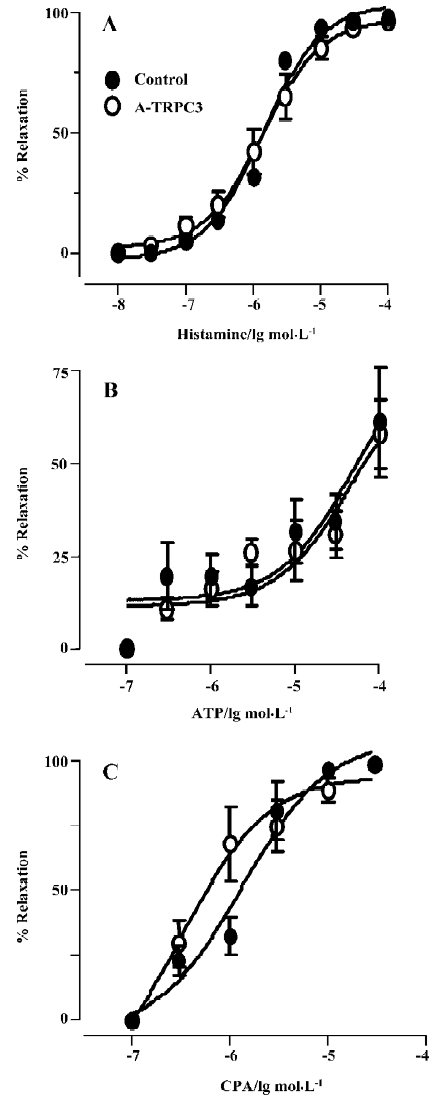
Effect of antisense oligo on agonist-induced relaxation The relaxation to common vasodilators including bradykinin, histamine, ATP and CPA was studied using a multi-myograph system. All these agonists elicited a concentration-dependent relaxation in rat small mesenteric arteries that were preconstricted with phenylephrine (0.5 µmol/L) plus U46619 (0.05 µmol/L). Two representative concentration-response curves for bradykinin in control oligo or antisense-TRPC3 treated arteries are shown in Figure 4A and 4B. While antisense-TRPC3 treatment did not show any effect on histamine-, ATP- and CPA-induced relaxation (Figure 5), it significantly suppressed the relaxation to bradykinin. In the antisense-TRPC3 group, the EC50 value of bradykinin was raised to 92.6±0.1 nmol/L from the control value of 32.6±0.1 nmol/L in the control group (Figure 4C). These data suggest that TRPC3 might participate in bradykinin-induced relaxation.
Effect of antisense oligo on flow-induced endothelial Ca2+ influx Flow-induced endothelial [Ca2+]i changes were compared between the arteries pretreated with antisense-TRPC3 or control oligos. In both types of arteries, flow initiated a transient rise in endothelial [Ca2+]i, which reached its peak in 20–30 s (Figure 6A). Treatment of the arteries with antisense-TRPC3 oligos significantly attenuated the magnitude of endothelial [Ca2+]i rise in response to flow (Figure 6B). The peak F1/F0 value was significantly decreased from 1.75±0.17 to 1.27±0.1.

Effect of antisense oligo on 30 nmol/L bradykinin-induced endothelial Ca2+ influx Because antisense-TRPC3 treatment significantly suppressed the relaxation to 30 nmol/L bradykinin, 30 nmol/L bradykinin-induced endothelial [Ca2+]i changes were compared between the arteries treated with antisense-TRPC3 and control oligos. Similar to flow responses, 30 nmol/L bradykinin induced a transient rise in endothelial [Ca2+]i in all tested arteries (Figure 6C). Treatment of the arteries with antisense-TRPC3 oligos significantly reduced the magnitude of endothelial [Ca2+]i rise in response to 30 nmol/L bradykinin (Figure 6D). The peak F1/F0 value was significantly decreased from 1.41±0.07 to 1.14±0.03. We also tested the effect of Gd3+ and nifedipine on bradykinin-induced Ca2+ influx. Gd3+ (10 µmol/L), a putative TRPC channel inhibitor, reduced the bradykinin-induced Ca2+ rise by 89%±2% (n=4), whereas an L-type Ca2+ channel blocker, nifedipine (1 µmol/L), had no effect (n=3).
Discussion
In the present study, we provided evidence that TRPC3 channel is involved in bradykinin- and flow-induced vasodilation in isolated rat small mesenteric arteries. In vessels isolated from antisense TRPC3 oligo-treated rats, the peak magnitude of flow-induced vasodilation was decreased by approximately 13% (Figure 3B). Antisense oligo treatment also reduced the sensitivity of vasodilation to bradykinin by increasing its EC50 value from the control value 32.6±0.1 nmol/L to 92.6±0.1 nmol/L (Figure 4), though the treatment does not appear to affect the maximal amplitude of relaxation to bradykinin. We also demonstrated that bradykinin- and flow-induced Ca2+ influx in the endothelial cells of isolated small mesenteric arteries was partly mediated by TRPC3. Further-more, antisense oligo treatment reduced flow- and bradykinin-induced Ca2+ influx in the endothelial cells of isolated small mesenteric arteries (Figure 6). Therefore, it is likely that bradykinin- and flow-induced vasodilation is partly attributed to the Ca2+ influx through TRPC3.
Evidence suggests that TRPC3 may participate in agonist-induced Ca2+ influx in endothelial cells in culture. In human vascular endothelial cells, expression of an N-terminal fragment of TRPC3, which exerts a dominant negative effect on the TRPC3 channel function, eliminated store-operated currents induced by thapsigargin or IP3, suggesting that TRPC3 channel plays a significant role in store-operated cation conductance[9]. Furthermore, bovine pulmonary artery endothelial cells transfected with TRPC3 channels display an increased Ca2+ influx in response to ATP and bradykinin[12]. These results suggest that TRPC3 forms agonist-activated Ca2+ permeable ion channels in endothelial cells in culture. Until now, however, there is still no evidence indicating the role of TRPC3 in endothelial Ca2+ influx and vasodilation in intact vessels. The results from the present study clearly indicated an involvement of TRPC3 in bradykinin- and flow-induced Ca2+ influx as well as vasodilation in isolated rat small mesenteric arteries. However, the effect of “knocking down” the expression of TRPC3 on bradykinin- and flow-induced vasodilation is relatively small. This could be because multiple TRPC isoforms may participate in endothelium-dependent dilation in rat mesenteric arteries. Previous data has demonstrated that TRPC4 is involved in acetylcholine-induced vasodilation in mice aorta[7]. Using immunohistochemical staining, we have found the expression of all TRPC proteins except TRPC7 (no commercial antibody available) in endothelial cells and smooth muscle cells of rat mesenteric arteries (Figure 2). In addition, it is possible that the expression of other TRPC channels may be upregulated for functional substitution when TRPC3 expression is suppressed.
Liu et al recently showed that TRPC3 channel expression was increased and this was responsible for an increased Ca2+ influx into monocytes in primary hypertension. More importantly, after specific TRPC3-knockdown using siRNA, the reduced TRPC3 expression in cells was accompanied by a significantly reduced Ca2+ influx[17]. In the present study, we have not only found an association between TRPC3 and bradykinin-induced Ca2+ influx, but also established a linkage between TRPC3 and bradykinin- and flow-induced vascular dilation. Therefore, it appears that TRPC3 is an important channel in the maintenance of normal vascular function, and that the overexpression of the channels could lead to the development of cardiovascular diseases.
CPA has been reported to be a specific inhibitor of Ca2+-dependent ATPase in the endoplasmic reticulum, which depletes the IP3-sensitive intracellular Ca2+ stores by blocking the refilling of Ca2+ stores. In the rat aorta, it may promote Ca2+ influx into endothelial cells to induce NO release from endothelial cells and then relax the aorta[18]. A different mechanism underlies the CPA-induced relaxation in mesenteric artery. Here CPA relaxes the mesenteric artery through the production of cAMP but not cGMP[11]. In the present study, we found that TRPC3 was not involved in the relaxation to CPA, ATP or histamine. It is unclear why TRPC3 is involved in dilation to bradykinin but not to CPA, ATP or histamine. One possibility is that different agonists may be coupled to different Ca2+ influx channels.
In the present study, immunohistochemical research clearly showed that the TRPC3 protein level was significantly reduced by the treatment of specific antisense oligo in endothelial cells as well as in smooth muscle cells (Figure 1). The suppression of TRPC3 expression in smooth muscle cells may change the basal vascular tone and/or alter the contractile response of the smooth muscle cells to the agonists that bind to smooth muscle cells, because TRPC is also a molecular component of Ca2+-permeable cation channels in smooth muscle cells. It has been demonstrated that the suppression of arterial TRPC3 expression in smooth muscle cells with antisense oligos significantly reduces the depolarization and constriction of intact cerebral arteries in response to UTP, not to increased intravascular pressure, that is, myogenic responses[21]. Nevertheless, because both bradykinin and flow first act on vascular endothelial cells to initiate vascular dilation, the suppression of smooth muscle TRPC3 is not expected to significantly alter the bradykinin- and flow-induced vasodilation.
In conclusion, with the use of antisense oligo, the present study directly demonstrated that TRPC3, as a Ca2+ influx channel, was involved in flow-induced vasodilation as well as in bradykinin-induced relaxation in rat small mesenteric arteries.
References
- Buchan KW, Martin W. Bradykinin induces elevations of cytosolic calcium through mobilisation of intracellular and extracellular pools in bovine aortic endothelial cells. Br J Pharmacol 1991;102:35-40.
- Busse R, Fleming I. Regulation of endothelium-derived vasoactive autacoid production by hemodynamic forces. Trends Pharmacol Sci 2003;24:24-9.
- Cooke JP, Rossitch E Jr, Andon NA, Loscalzo J, Dzau VJ. Flow activates an endothelial potassium channel to release an endogenous nitrovasodilator. J Clin Invest 1991;88:1663-71.
- Davies PF. Flow-mediated endothelial mechanotransduction. Physiol Rev 1995;75:519-60.
- Falcone JC, Kuo L, Meininger GA. Endothelial cell calcium increases during flow-induced dilation in isolated arterioles. Am J Physiol 1993;264:H653-H659.
- Freay A, Johns A, Adams DJ, Ryan US, Van Breemen C. Bradykinin and inositol 1,4,5-trisphosphate-stimulated calcium release from intracellular stores in cultured bovine endothelial cells. Pflugers Arch 1989;414:377-84.
- Freichel M, Suh SH, Pfeifer A, Schweig U, Trost C, Weissgerber P, et al. Lack of an endothelial store-operated Ca2+ current impairs agonist-dependent vasorelaxation in TRP4-/- mice. Nat Cell Biol 2001;3:121-7.
- Gao X, Wu L, O’Neil RG. Temperature-modulated diversity of TRPV4 channel gating: activation by physical stresses and phorbol ester derivatives through protein kinase C-dependent and -independent pathways. J Biol Chem 2003;278:27129-37.
- Groschner K, Hingel S, Lintschinger B, Balzer M, Romanin C, Zhu X, et al. Trp proteins form store-operated cation channels in human vascular endothelial cells. FEBS Lett 1998;437:101-6.
- Huang CL. The transient receptor potential superfamily of ion channels. J Am Soc Nephrol 2004;15:1690-9.
- Kamata K, Umeda F, Kasuya Y. Possible existence of novel endothelium-derived relaxing factor in the endothelium of rat mesenteric arterial bed. J Cardiovasc Pharmacol 1996;27:601-6.
- Kamouchi M, Philipp S, Flockerzi V, Wissenbach U, Mamin A, Raeymaekers L, et al. Properties of heterologously expressed hTRP3 channels in bovine pulmonary artery endothelial cells. J Physiol 1999;518:345-58.
- Kohler R, Brakemeier S, Kuhn M, Degenhardt C, Buhr H, Pries A, et al. Expression of ryanodine receptor type 3 and TRP channels in endothelial cells: comparison of in situ and cultured human endothelial cells. Cardiovasc Res 2001;51:160-8.
- Kwan HY, Huang Y, Yao X. Regulation of canonical transient receptor potential isoform 3 (TRPC3) channel by protein kinase G. Proc Natl Acad Sci USA 2004;101:2625-30.
- Lin MJ, Leung GP, Zhang WM, Yang XR, Yip KP, Tse CM, et al. Chronic hypoxia-induced upregulation of store-operated and receptor-operated Ca2+ channels in pulmonary arterial smooth muscle cells: a novel mechanism of hypoxic pulmonary hyper-tension. Circ Res 2004;95:496-505.
- Liu C, Mather S, Huang Y, Garland CJ, Yao X. Extracellular ATP facilitates flow-induced vasodilatation in rat small mesenteric arteries. Am J Physiol Heart Circ Physiol 2004;286:H1688-H1695.
- Liu D, Scholze A, Zhu Z, Kreutz R, Wehland-von-Trebra M, Zidek W, et al. Increased transient receptor potential channel TRPC3 expression in spontaneously hypertensive rats. Am J Hypertens 2005;18:1503-7.
- Moritoki H, Hisayama T, Takeuchi S, Kondoh W, Imagawa M. Relaxation of rat thoracic aorta induced by the Ca2+-ATPase inhibitor, cyclopiazonic acid, possibly through nitric oxide formation. Br J Pharmacol 1994;111:655-42.
- Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet 2003;33:129-37.
- Nilius B, Droogmans G. Ion channels and their functional role in vascular endothelium. Physiol Rev 2001;81:1415-59.
- Reading SA, Earley S, Waldron BJ, Welsh DG, Brayden JE. TRPC3 mediates pyrimidine receptor-induced depolarization of cerebral arteries. Am J Physiol Heart Circ Physiol 2005;288:H2055-H2061.
- Thebault S, Zholos A, Enfissi A, Slomianny C, Dewailly E, Roudbaraki M, et al. Receptor-operated Ca2+ entry mediated by TRPC3/TRPC6 proteins in rat prostate smooth muscle (PS1) cell line. J Cell Physiol 2005;204:320-8.
- Tiruppathi C, Freichel M, Vogel SM, Paria BC, Mehta D, Flockerzi V, et al. Impairment of store-operated Ca2+ entry in TRPC4(-/-) mice interferes with increase in lung microvascular permeability. Circ Res 2002;91:70-6.
- Yamamoto K, Korenaga R, Kamiya A, Ando J. Fluid shear stress activates Ca2+ influx into human endothelial cells via P2X4 purinoceptors. Circ Res 2000;87:385-91.
- Yamamoto K, Sokabe T, Ohura N, Nakatsuka H, Kamiya A, Ando J. Endogenously released ATP mediates shear stress-induced Ca2+ influx into pulmonary artery endothelial cells. Am J Physiol Heart Circ Physiol 2003;285:H793-H803.
- Yao X, Garland CJ. Recent developments in vascular endothelial cell transient receptor potential channels. Circ Res 2005;97:853-63.
- Yao X, Kwan HY, Chan FL, Chan NW, Huang Y. A protein kinase G-sensitive channel mediates flow-induced Ca2+ entry into vascular endothelial cells. FASEB J 2000;14:932-8.