
SRC-3/AIB1: transcriptional coactivator in oncogenesis1
Introduction
Steroid receptor coactivator-3 (SRC-3), localized in a frequently amplified chromosomal region, 20q12[1], was first identified as Amplified in Breast cancer 1 (AIB1), also known as NCoA3, p/CIP[2], RAC3[3], ACTR[4], and TRAM1[5]. Biochemical and cellular biological analyses revealed that SRC-3 belongs to the p160 steroid receptor coactivator family, which includes SRC-1 and SRC-2 (TIF2/GRIP1)[6]. SRC interact not only with nuclear receptors, such as estrogen receptor (ER)[7], progesterone receptor (PR)[7], and thyroid receptor[5,8], but also with other transcription factors, including activator protein-1 (AP-1)[9], nuclear factor-κB (NFκB)[10], signal transducer and activator of transcription (STAT)[11] and E2F1[12]. Binding of SRC to transcription factors will further recruit other chromatin modification factors, such as acetyltransferases (CBP and p300) and methyltransferases (CARM1 and PRMT1), modify the chromatin structure and activate transcription of their target genes[13,14]. Thus, it is conceivable that changes of their concentrations and activities may greatly affect the expression levels of many genes and, as a consequence, influence a variety of cellular processes.
Recently, SRC-3 has received more attention, because a growing body of evidence has revealed that overexpression of SRC-3 might promote initiation and/or progression of carcinogenesis by affecting many important signal pathways. In this review, we will focus on the involvement of SRC-3 in oncogenesis and discuss the potential mechanisms by which this occurs.
Molecular structure of SRC-3
The SRC-3 gene encodes a 160 kDa coactivator, with 40% sequence similarity to other SRC family members. Furthermore, SRC family members contain multiple similar functional domains (Figure 1). Their N-terminal basic helix-loop-helix-Per/ARNT/Sim (bHLH-PAS) domain is the most conserved region among SRC family members (60% identity for amino acids 1–350)[1]. The bHLH-PAS domain can serve as DNA-binding, protein–protein interaction surfaces for various bHLH-PAS-containing factors[15]. Actually, SRC family members have been shown to interact with myogenin, MEF-2C and transcriptional enhancer factor (TEF) through this domain[16,17]. It is also possible that this region could participate in intramolecular interactions to regulate the activity of p160 coactivators or intermolecular interactions with other coactivators. One example is coiled-coil coactivator (CoCoA), which binds the bHLH-PAS domain of SRC-2 and enhances ER-mediated transcription[18].
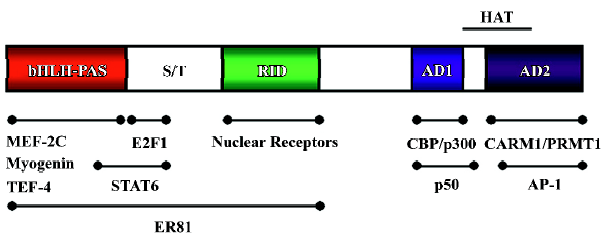
The conserved central region of the SRC contains multiple LXXLL motifs (where L is leucine and X is any amino acid), which interact with a hydrophobic cleft in the nuclear receptor LDB formed as a result of ligand-induced conformational changes[19,20]. Two intrinsic transcriptional activation domains (AD1 and AD2) are located at the C-terminal of the receptor interaction domain of the SRC molecules. The AD1 contains multiple LXXLL motifs that are responsible for interactions with the histone acetyltransferase (HAT) CBP and p300. Although C-terminal domains of SRC-1 and SRC-3 possess HAT activity, its HAT activities are weaker than those of CBP, p300 and p/CAF[21]. Therefore, the importance of SRC HAT activity remains unclear. AD2 can interact with protein arginine methyltransferases (PRMT), such as CARM1 and PRMT1[22,23]. Based on the protein structure, it has been suggested that SRCs serve as adaptor proteins to recruit additional coactivators and basal transcriptional machinery onto the promoter. Such recruitment may be critical for nuclear receptor-directed local chromatin remodeling and assembly of the transcriptional machinery around the promoter.
SRC-3 in cancer
Clinical study SRC-3 is a steroid receptor coactivator, and SRC-3 amplification and/or overexpression is detected in many hormone-sensitive tumors, such as breast, prostate, and ovarian cancer, and meningioma[1,24–27]. In breast cancer biopsies, SRC-3 was shown to be amplified and overexpress-ed in 5%–10% and 30%–60% of cases, respectively[1,28]. In tamoxifen-treated breast cancer patients, SRC-3 overexpression is associated with high levels of HER-2/neu, tamoxifen resistance, and poor disease-free survival, suggesting that cross-talk between SRC-3, HER2/neu and ER signal pathways is important in breast cancer[30,31]. In the case of prostate cancer, a study of SRC-3 expression in a series of 37 patients revealed that its expression level correlated significantly with tumor grade and stage of disease[24]. Further analysis of a cohort of 480 patients with prostate cancer revealed that overexpression of SRC-3 was correlated with tumor recurrence and survival[25]. Moreover, it has been reported that a splicing variant of SRC-3 (SRC-3-Δ3), encoding a 130 kDa protein that lacks the N-terminal bHLH and a portion of the PAS domain, is overexpressed in breast cancer biopsies. SRC-3-Δ3 is a highly potent steroid receptor coactivator in vitro, even when present at a relatively low level, as compared with the full-length SRC-3[32]. These data suggest that SRC-3 plays an essential role in hormone-sensitive tumors, probably by activating the activity of the steroid receptor. Intriguingly, a recent study has revealed that SRC-3 overexpression is correlated with the absence of estrogen and progesterone receptors in breast cancer[29]. This suggests that SRC-3 may also function via other transcription factor(s) during tumorigenesis.
Consistent with this finding, SRC-3 is also found to be involved in many types of non-steroid-targeted tumors, such as pancreatic cancer, gastric cancer, colorectal carcinoma and hepatocellular carcinoma (HCC)[33–37]. An increased number of SRC-3 gene copies were detected in 37% of pancreatic adenocarcinoma cases[34]. Furthermore, progressive increased frequency of SRC-3 expression was detected during pancreas cancer progression (the SRC-3 expression level follows the following order: pancreatitis < low-grade pancreatic intraepithelial neoplasia [PanIN] < high-grade PanIN < invasive ductal adenocarcinomas). Moreover, SRC-3 is also closely associated with metastasis and tumor recurrence in gastric cancer[35] and HCC[37]. Taken together, these clinical data suggest that SRC-3 may play an important role in the genesis of human cancers in a hormone-independent manner.
Lessons from mouse models
Transgenic mouse model To determine whether SRC-3 is a bona fide oncogene, an SRC-3 transgenic mouse model was generated by M Brown and colleagues[38]. The over-expression of SRC-3, under the control of mouse mammary tumor virus (MMTV) LTR in transgenic mice, was associated with an extremely high tumor incidence (76%) in aging animals, with an average latency of 16 months. High tumor incidence was found in mammary glands (48/145 detected tumors), pituitary (42/145), uterus (18/145) and lung (18/145), consistent with the high ectopic expression of SRC-3 in these organs[38]. Further analysis revealed that the persistent hyperplastic lesions were caused by a combination of increased cellular proliferation and reduced apoptosis. IGF-1, a growth factor important for cancer cell growth and survival, was induced at both the mRNA and protein levels in normal mammary gland of SRC-3 transgenic mice. The serum level of IGF-1 was also elevated in transgenic mice, suggesting that IGF-1 signaling is essential for SRC-3-induced tumorigenesis. Moreover, most of the mammary tumors were invasive, and several adenocarcinomas were metastatic. Finally, the genesis of mammary tumors is independent of the formation of pituitary adenomas, the reproductive history of the mice and the ER status, suggesting that SRC-3 induced mammary tumors are not dependent on hormone level.
Because SRC-3-Δ3, a splicing variant of SRC-3, was reported to be overexpressed in breast cancer specimens, overexpression of SRC-3-Δ3, under the control of the cytome-galovirus (CMV) promoter in transgenic mice, was generated by AT Riegel and colleagues[39]. Mammary epithelial cell proliferation and ductal ectasia were found in CMV-SRC-3-Δ3 mice, which is similar to the phenotype found in MMTV-SRC-3 mice[38]. Moreover, both mouse models showed that prolactin, a hormone produced in the pituitary, was not related to SRC-3- or SRC-3-Δ3-induced cell proliferation, which indicates that elevated hyperplasia is not related to prolactin. However, no tumor was found in CMV-SRC-3-Δ3 animals, probably due to the lower transgene expression levels (no more than two-fold) and the lack of elevated systemic IGF-1 levels[39]. These results indicate that both SRC-3 splicing variants are capable of inducing mammary epithelial cell proliferation. Therefore, MMTV-SRC-3 transgenic mouse model provides evidence that SRC-3 is an oncogene and that its overexpression is sufficient to trigger the initiation of tumorigenesis, which ultimately leads to invasive carcinoma.
Knockout mouse model SRC-3 knockout mice have retarded growth, reduction in mammary gland alveolar development during pregnancy, and resistance to growth hormones and estrogen[40,41]. In addition, SRC-3 deficiency significantly suppresses the incidence of MMTV-v-Ha-ras oncogene-induced mammary gland ductal hyperplasia, tumorigenesis, and metastasis to the lung[42]. Most SRC-3+/+-ras and SRC-3+/–-ras mice developed many mammary intraepithelial neoplasia lesions by age 17 weeks. Fifty percent of the SRC3+/+-ras mice (n=38) and SRC3+/–-ras mice (n=46) developed breast tumors by age 32.5 and 42 weeks, respectively. All of these mice developed palpable breast tumors by age 70 weeks. In comparison, approximately 50% of the SRC-3–/–-ras virgin mice still had normal mammary gland morphology by age 80 weeks. Significant differences in mammary tumor incidence were also observed between SRC-3+/+-ras and SRC-3–/–-ras mice under different hormonal conditions, suggesting that SRC-3-induced tumorigenesis is not likely through hormone regulated events.
Moreover, depletion of SRC-3 also significantly and selectively suppresses the mammary tumorigenesis induced by chemical carcinogen 7,12-dimethylbenz[a]-anthracene (DMBA) with or without pituitary isografts[43]. Mammary tumor incidence dropped from 45% and 44% in DMBA-treated SRC-3+/+ and SRC-3+/– mice, respectively, to 11% in DMBA-treated SRC-3–/– mice. Although skin tumors were also detected in these three groups, there were no significant differences in skin tumor frequencies.
Molecular mechanisms of SRC-3 function
Hormone-dependent signal transduction pathway Hormone modulation is closely related to tumorigenesis in breast, ovarian and prostate cancer. SRC-3 is a member of the steroid receptor coactivator family and is necessary for the complete functioning of ER[44,45]. Therefore, it is likely that SRC-3 also plays an important role in estrogen-stimulated proliferation of breast tumors. In line with this notion, suppression of SRC-3 leads to reduction of recruitment of ERα and polymerase II to its target gene promoter, resulting in the inhibition of transcription. Interestingly, inhibition of SRC-3 protein expression has a more detrimental effect on ERα target gene regulation than does inhibition of the other p160 protein, SRC-1[46]. This result suggests possible coactivator specificity and SRC-3 may have more impact on ER activity than other members. Cyclin D1, which is frequently overexpressed in tumors, is an ERα target gene, and its expression is enhanced by SRC-3 through functional interaction of the estrogen receptor with the cyclin D1 promoter[47]. SRC-3-Δ3, the splicing variant of SRC-3, can increase the estrogenicity of a variety of natural and pharmacologic compounds in tissues that develop hormone-dependent neoplasia. Thus, overexpression of SRC-3-Δ3 may be a contributing factor to the development of hormone-driven neoplasia and to hormone resistant breast cancers[48]. Taken together, these findings indicate that SRC-3 is required for maximal activity of ER and other hormone receptors. Over-expression and/or amplification of SRC-3 is likely to facilitate transformation by ER signaling in breast cancer.
Hormone-independent signal transduction pathway SRC -3 abnormality has been detected in tumors that are not targeted by steroid hormones, such as gastric cancer and HCC[35–37]. Overexpression of SRC-3 also exists in ER- or PR-negative breast cancer[29]. This clinical evidence strongly supports the hypothesis that SRC-3 can enhance hormone-independent proliferation and survival during tumorigenesis. Extensive investigations reveal that SRC-3 can interact with a broad spectrum of transcription factors in addition to hormone receptors[9–12]. Hence, many signal pathways, other than hormone receptors, can be affected by overexpression of SRC-3 in cancer cells. Deregulation of this signaling will facilitate tumorigenesis by altering cell differentiation, proliferation, survival and metastasis.
E2F1 signal pathway Regulation of cell cycle progression in mammalian cells is very complex, involving many signal molecules. Among them, E2F1 transcription factor has been shown to regulate cell cycle progression by modulating the expression of proteins required for the G1/S transition and DNA synthesis. In the G0 and early G1 phases, Rb binds to E2F1 and suppresses the transcriptional activity of E2F1. Phosphorylation of Rb family proteins by cyclin-dependent kinase (CDK) results in the release of E2F1[49]. The freed E2F1, which is present as a heterodimer with its binding partner DP-1 or DP-2, is then activated and promotes the transcription of its target genes, such as cyclin A and cyclin E. Because E2F1 is an important regulator of cell cycle progression, it is not surprising that transcription activity of E2F1 is frequently induced in cancer cells. E2F4, another E2F family member, appears to function as a repressor by recruiting Rb family proteins to E2F-regulated promoters. The repressor E2F4 is believed to be required for cell cycle exit and differentiation[50].
Recently, SRC-3 has been found to directly interact with E2F1, but not with E2F4[12]. The structural difference at the N-terminal of E2F1 and E2F4 may account for this differential binding[50]. The elevated expression of SRC-3 may expand the available pool of the SRC-3-E2F1 complexes in quiescent cells to displace the repressive E2F complex from E2F1-responsive promoters. Consequently, SRC-3-E2F1 trans-activates the E2F1 target genes involved in cell proliferation. Those genes include cyclin E, Cdk2, cyclin A, cdc25A and E2F1 itself. Both E2F1 and cyclin E are potential proliferative markers, and are closely correlated with poor outcomes of breast cancer[51–53]. Furthermore, ectopic expression of cyclin E in E2-responsive cells can effectively overcome the growth arrest effected by antiestrogens[54]. Hence, SRC-3-induced E2F1 signaling may be an important mechanism for hormone-independent breast cancer cell proliferation, as well as for other non-hormone dependent cancers.
Insulin-like growth factor-1/AKT signal pathway Insulin-like growth factor-1 (IGF-1)/AKT signaling has diverse roles in cell processes such as cell growth, proliferation, survival, and migration[55]. The binding of IGF-1 to its cognate receptor, IGF1R, triggers phosphorylation of insulin receptor substrates (IRS-1 and IRS-2) and activation of phos-phatidylinositol 3-kinase (PI3K), which is followed by activation of AKT (Figure 2). Both in vitro and in vivo data reveal that the SRC-3 expression level is closely and positively associated with the IGF-1 expression level[38,40,41]. Moreover, the IGF1Rα protein level is reduced in SRC-3 knockdown breast cancer cells, whereas IGF1Rα is induced in CMV-SRC-3-Δ3 transgenic mice. Furthermore, IGF1Rα is highly phosphorylated in tumors derived from MMTV-SRC-3 transgenic mice[38,39,56]. Interestingly, no significant difference in IGF1Rβ was found, despite insulin response substrate-1 (IRS-1) and IRS-2 being dramatically suppressed in SRC-3–/–-Ras transgenic mice. As expected, IGF-1 downstream effector, AKT expression level and activity become elevated on overexpression of SRC-3 in a prostate cancer cell line[57]. Consistently, the activity of AKT is increased in a MMTV-SRC-3 mouse model[38].
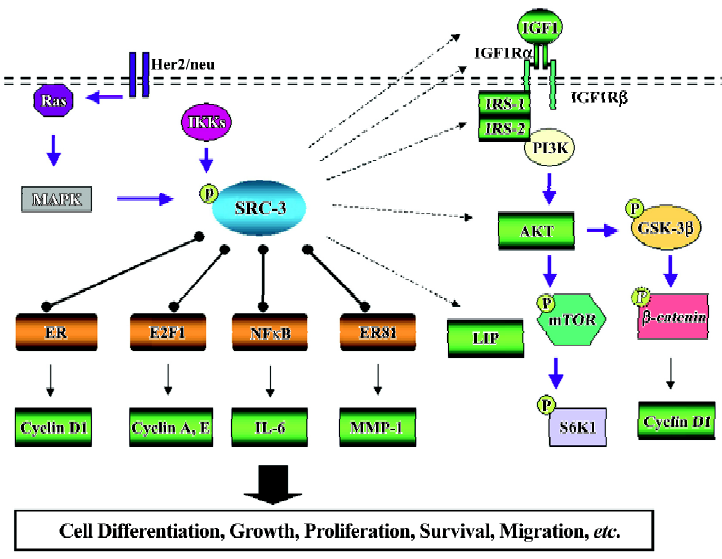
As a result, multiple downstream pathways of AKT signaling are affected, mostly at the phosphorylation level. SRC-3 overexpression enhances somatic cell growth in prostate cancer cells by activating AKT/mTOR signaling[57]. In a MMTV-SRC-3 transgenic mice model, GSK-3β, the substrate of AKT, is strongly phosphorylated at Ser 9, resulting in a reduced β-catenin phosphorylation level (Ser 45). Unphos-phorylated β-catenin will evade proteinase degradation and translocate into the nucleus, leading to the activation of target genes, such as cyclin D1[38] (Figure 2). Collectively, the data above indicate that the expression level and/or activity of many components in the IGF/AKT signaling are under the strict control of SRC-3.
NF-κB signal pathway Rel/Nuclear factor-κB (NF-κB) is a dimeric transcription factor that plays important roles in the control of growth, differentiation, and apoptosis[58]. Rel/NF-κB consists of homodimers and heterodimers formed by several subunits: NF-κB1 (p50/p105), NF-κB2 (p52/p100), RelA (p65), Rel B, and c-Rel proteins. The inactive form of NF-κB is localized in the cytoplasm and consists of 3 subunits: the DNA-binding p50 and p65 subunits, and an inhibitory subunit, called IκB, which is bound to p65. Once IκB is released through phosphorylation by IκB kinase (IKK), NF-κB will translocate to the nucleus and bind to target sites on DNA[58]. Activation of NF-κB results in the induction of a large number of genes that influence cellular proliferation, inflammation, and cellular adhesion[58]. Furthermore, aberrant NF-κB activation has been implicated in the pathogenesis of several human malignancies, including cancer of the breast, prostate, gastrointestinal tract, liver, pancreas and skin.
SRC-3 can interact and coactivate p65/NFκB[59]. Recently, SRC-3, but not SRC-1, has been reported to associate with IKK, suggesting the essential role of SRC-3 in the NF-κB signaling pathway[60]. In response to tumor necrosis factor (TNF)-α, SRC-3 is phosphorylated by the IKK complex. As a result, SRC-3 and NFκB translocate from the cytosol to the nucleus. One of the putative target genes of SRC-3, interleukin (IL)-6, which has an NF-κB binding site in its promoter and plays an important role in tumor metastasis and inflammation[61], is induced. Elevated IL-6 in both serum and prostate cancer tissues acts as an autocrine growth factor in prostate cancer progression[62,63]. Consistent with this notion, introduction of wild type SRC-3 in SRC-3 null MEF cells can restore IL-6 induction by TNF-α, but not SRC-3 phosphorylation mutants[64]. This result indicates that phosphorylated SRC-3 is tightly linked to NF-κB transcriptional activity. For a more detailed discussion of the phosphorylation of SRC-3, readers are referred to a recent review by Wu et al[65].
C/EBPβ signal pathway The C/EBPβ family of transcription factors has been implicated in the regulation of proliferation and differentiation in the mammary gland and breast carcinogenesis[66,67]. There are 2 C/EBPβ isoforms: liver-enriched activating protein (LAP) and liver-enriched inhibitory protein (LIP). In comparison with LAP, LIP lacks the N-terminal transactivation domain, but retains the dimerization and DNA-binding domain; therefore, it is thought that LIP functions as a dominant negative C/EBPβ isoform. In the mammary gland, increased LIP expression is associated with rapid mammary epithelial cell proliferation during pregnancy[66].
In CMV-SRC-3-Δ3 transgenic mice, there is a reduced LAP/LIP ratio, due to the induction of LIP expression levels[39]. Reduction of the LAP/LIP ratio by LIP overexpression in a mouse model can trigger hyperplasia in an ER-independent manner, suggesting that an increase in LIP levels is related to mammary ductal hyperplasia in CMV-SRC-3-Δ3 transgenic mice. However, so far little is known about how overexpres-sion of SRC-3 can preferentially induce LIP expression. Further study is required to investigate whether SRC-3 induces LIP directly at the transcriptional level or at the translational level, and whether SRC-3 can directly bind to the C/EBPβ promoter.
HER2/neu/MAPK signal pathway Although deregulated SRC-3 does not affect MAPK pathway activity in cells or in a mouse model[43,57], SRC-3 serves as a conduit from MAPK signaling to ER (Figure 2). MAPK can regulate SRC-3 activity by phosphorylating SRC-3 and augment ER activity by increasing recruitment of p300 and associated histone acetyltransferase activity[68]. Recently, a clinical study on breast cancer patients showed that SRC-3 and HER2/neu expression levels are closely associated with the development of tamoxifen resistance, suggesting that the crosstalk between HER2/neu and SRC-3 exists[30]. Further study indicated that tamoxifen recruits coactivator complexes to the ER-regulated pS2 gene promoter in SRC-3 and HER2 over-expressing cells, whereas tamoxifen recruits corepressor complexes in SRC-3 overexpressing cells without the overexpres-sion of HER2. It is proposed that the switch from corepressor complexes to coactivator complexes might be the reason for the loss of tamoxifen resistance[31]. Importantly, treatment with gefitinib, a factor inhibiting crosstalk of EGFR and HER2 with ER, significantly restores tamoxifen’s antagonistic effect on gene expression and anti-tumor function.
Concluding remarks and future perspectives As illustrated above, more and more compelling evidence reveals that SRC-3 is a bona fide oncogene. First of all, use of a MMTV-SRC-3 transgenic mouse model clearly indicates that full-length SRC-3 is sufficient to initiate tumorigenesis. Furthermore, SRC-3 overexpression exists in a variety of cancer types and integrates several vital signal pathways, such as nuclear receptor, HER2/neu, IGF/AKT, and NFκB, which are frequently deregulated in many cancers. These signal pathways are involved in cell proliferation, survival and migra-tion, suggesting the central role of SRC-3 in tumorigenesis.
Reduction of SRC-3 in cancer cells by RNAi knockdown or ribozymes can markedly reduce tumor formation in nude mice[25,45]; and a deficiency of SRC-3 in a mouse model can suppress tumorigenesis, when challenged with a carcinogen or the oncogene H-ras[42,43]. Therefore, SRC-3 per se is a very attractive chemotherapeutic target.
However, there are still a number of important questions that remain to be addressed. Because of the role of SRC-3 in carcinogenesis, it is worth asking how the activities of SRC-3 are modulated during carcinogenesis. Because elevated SRC-3 can selectively activate one subset of nuclear receptors and transcription factors, such as E2F1, the selectivity of SRC-3 function in a cancer context can lead to the finding of new drug targets and novel therapeutic strategies. It has been suggested that phosphorylated SRC-3 is associated with its oncogenic potential; thus, it might be important to evaluate phosphorylated SRC-3 and/or investigate whether there are any activated mutations in SRC-3 in tumor biopsies. Recently SRC-3 was found to physically interact with ER81, a PEA3 family member, and drive MMP-1 expression, which might be involved in metastasis[69]. Thus, it is not unlikely that SRC-3 interacts with other transcription factors or even other proteins unrelated to the transcription complex. Examination of the SRC-3 complex in a cancer context might enhance our understanding of the role of SRC-3 in carcinogenesis.
Acknowledgement
We thank Dr Ray-chang WU and Dr Khoi CHU for their critical comments.
References
- Anzick SL, Kononen J, Walker RL, Azorsa DO, Tanner MM, Guan XY, et al. AIB1, a novel estrogen receptor co-activator amplified in breast and ovarian cancer. Science 1997;277:965-8.
- Torchia J, Rose DW, Inostroza J, Kamei Y, Westin S, Glass CK, et al. The transcriptional coactivator p/CIP binds CBP and mediates nuclear receptor function. Nature 1997;387:677-84.
- Li H, Gomes PJ, Chen JD. RAC3 a steroid/nuclear receptor-associated coactivator that is related to SRC-1 and TIF2. Proc Natl Acad Sci USA 1997;94:8479-84.
- Chen H, Lin RJ, Schiltz RL, Chakravarti D, Nash A, Nagy L, et al. Nuclear receptor coactivator ACTR is a novel histone acetyl-transferase and forms a multimeric activation complex with p/CAF and CBP/p300. Cell 1997;90:569-80.
- Takeshita A, Cardona GR, Koibuchi N, Suen CS, Chin WW. TRAM-1, a novel 160 kDa thyroid hormone receptor activator molecule, exhibits distinct properties from steroid receptor coactivator-1. J Biol Chem 1997;272:27629-34.
- Suen CS, Berrodin TJ, Mastroeni R, Cheskis BJ, Lyttle CR, Frail DE. A transcriptional coactivator, steroid receptor coactivator-3, selectively augments steroid receptor transcriptional activity. J Biol Chem 1998;273:27645-53.
- Han SJ, Demayo FJ, Xu J, Tsai SY, Tsai MJ, O’Malley BW. Steroid receptor coactivators SRC-1 and SRC-3 differentially modulate tissue-specific activation functions of the progesterone receptor. Mol Endocrinol 2006;20:45-55.
- Ying H, Furuya F, Willingham MC, Xu J, O’Malley BW, Cheng SY. Dual functions of the steroid hormone receptor coactivator 3 in modulating resistance to thyroid hormone. Mol Cell Biol 2005;25:7687-95.
- Lee SK, Kim HJ, Na SY, Kim TS, Choi HS, Im SY, et al. Steroid receptor coactivator-1 coactivates activating protein-1-mediated transactivations through interaction with the c-Jun and c-Fos subunits. J Biol Chem 1998;273:16651-4.
- Werbajh S, Nojek I, Lanz R, Costas MA. RAC-3 is a NF-kappa B coactivator. FEBS Lett 2000;485:195-9.
- Arimura A, van Peer M, Schroder AJ, Rothman PB. The transcriptional co-activator p/CIP (NCoA-3) is up-regulated by STAT6 and serves as a positive regulator of transcriptional activation by STAT6. J Biol Chem 2004;279:31105-12.
- Louie MC, Zou JX, Rabinovich A, Chen HW. ACTR/AIB1 functions as an E2F1 coactivator to promote breast cancer cell proliferation and antiestrogen resistance. Mol Cell Biol 2004;24:5157-71.
- Chen D, Ma H, Hong H, Koh SS, Huang SM, Schurter BT, et al. Regulation of transcription by a protein methyltransferase. Science 1999;284:2174-7.
- McKenna NJ, O’Malley BW. Combinatorial control of gene expression by nuclear receptors and coregulators. Cell 2002;108:465-74.
- Huang ZJ, Edery I, Rosbash M. PAS is a dimerization domain common to Drosophila period and several transcription factors. Nature 1993;364:259-62.
- Belandia B, Parker MG. Functional interaction between the p160 coactivator proteins and the transcriptional enhancer factor family of transcription factors. J Biol Chem 2000;275:30801-5.
- Chen SL, Dowhan DH, Hosking BM, Muscat GE. The steroid receptor coactivator, GRIP-1, is necessary for MEF-2C-dependent gene expression and skeletal muscle differentiation. Genes Dev 2000;14:1209-28.
- Kim JH, Li H, Stallcup MR. CoCoA, a nuclear receptor coactivator which acts through an N-terminal activation domain of p160 coactivators. Mol Cell 2003;12:1537-49.
- Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, et al. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 1998;95:927-37.
- Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engstrom O, et al. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 1997;389:753-8.
- Spencer TE, Jenster G, Burcin MM, Allis CD, Zhou J, Mizzen CA, et al. Steroid receptor coactivator-1 is a histone acetyltrans-ferase. Nature 1997;389:194-8.
- Chen D, Ma H, Hong H, Koh SS, Huang SM, Schurter BT, et al. Regulation of transcription by a protein methyltransferase. Science 1999;284:2174-7.
- Koh SS, Chen D, Lee YH, Stallcup MR. Synergistic enhancement of nuclear receptor function by p160 coactivators and two coactivators with protein methyltransferase activities. J Biol Chem 2001;276:1089-98.
- Gnanapragasam VJ, Leung HY, Pulimood AS, Neal DE, Robson CN. Expression of RAC 3, a steroid hormone receptor co-activator in prostate cancer. Br J Cancer 2001;85:1928-36.
- Zhou HJ, Yan J, Luo W, Ayala G, Lin SH, Erdem H, et al. SRC-3 is required for prostate cancer cell proliferation and survival. Cancer Res 2005;65:7976-83.
- Tanner MM, Grenman S, Koul A, Johannsson O, Meltzer P, Pejovic T, et al. Frequent amplification of chromosomal region 20q12-q13 in ovarian cancer. Clin Cancer Res 2000;6:1833-9.
- Carroll RS, Brown M, Zhang J, DiRenzo J, De Mora JF, Black PM. Expression of a subset of steroid receptor cofactors is asso-ciated with progesterone receptor expression in meningiomas. Clin Cancer Res 2000;6:3570-5.
- List HJ, Reiter R, Singh B, Wellstein A, Riegel AT. Expression of the nuclear coactivator AIB1 in normal and malignant breast tissue. Breast Cancer Res Treat 2001;68:21-8.
- Bouras T, Southey MC, Venter DJ. Overexpression of the steroid receptor coactivator AIB1 in breast cancer correlates with the absence of estrogen and progesterone receptors and positivity for p53 and HER2/neu. Cancer Res 2001;61:903-7.
- Osborne CK, Bardou V, Hopp TA, Chamness GC, Hilsenbeck SG, Fuqua SA, et al. Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J Natl Cancer Inst 2003;95:353-61.
- Shou J, Massarweh S, Osborne CK, Wakeling AE, Ali S, Weiss H, et al. Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J Natl Cancer Inst 2004;96:926-35.
- Reiter R, Wellstein A, Riegel AT. An isoform of the coactivator AIB1 that increases hormone and growth factor sensitivity is overexpressed in breast cancer. J Biol Chem 2001;276:39736-41.
- Henke RT, Haddad BR, Kim SE, Rone JD, Mani A, Jessup JM, et al. Overexpression of the nuclear receptor coactivator AIB1 (SRC-3) during progression of pancreatic adenocarcinoma. Clin Cancer Res 2004;10:6134-42.
- Ghadimi BM, Schrock E, Walker RL, Wangsa D, Jauho A, et al. Specific chromosomal aberrations and amplification of the AIB1 nuclear receptor coactivator gene in pancreatic carcinomas. Am J Pathol 1999;154:525-36.
- Sakakura C, Hagiwara A, Yasuoka R, Fujita Y, Nakanishi M, Masuda K, et al. Amplification and over-expression of the AIB1 nuclear receptor co-activator gene in primary gastric cancers. Int J Cancer 2000;89:217-23.
- Xie D, Sham JS, Zeng WF, Lin HL, Bi J, Che LH, et al. Correlation of AIB1 overexpression with advanced clinical stage of human colorectal carcinoma. Hum Pathol 2005;36:777-83.
- Wang Y, Wu MC, Sham JS, Zhang W, Wu WQ, Guan XY. Prognostic significance of c-myc and AIB1 amplification in hepatocellular carcinoma. A broad survey using high-throughput tissue microarray. Cancer 2002;95:2346-52.
- Torres-Arzayus MI, De Mora JF, Yuan J, Vazquez F, Bronson R, Rue M, et al. High tumor incidence and activation of the PI3K/AKT pathway in transgenic mice define AIB1 as an oncogene. Cancer Cell 2004;6:263-74.
- Tilli MT, Reiter R, Oh AS, Henke RT, McDonnell K, Gallicano GI, et al. Overexpression of an N-terminally truncated isoform of the nuclear receptor coactivator amplified in breast cancer 1 leads to altered proliferation of mammary epithelial cells in transgenic mice. Mol Endocrinol 2005;19:644-56.
- Xu J, Liao L, Ning G, Yoshida-Komiya H, Deng C, O’Malley BW. The steroid receptor coactivator SRC-3 (p/CIP/RAC3/AIB1/ACTR/TRAM-1) is required for normal growth, puberty, female reproductive function, and mammary gland development. Proc Natl Acad Sci USA 2000;97:6379-84.
- Wang Z, Rose DW, Hermanson O, Liu F, Herman T, Wu W, et al. Regulation of somatic growth by the p160 coactivator p/CIP. Proc Natl Acad Sci USA 2000;97:13549-54.
- Kuang SQ, Liao L, Zhang H, Lee AV, O’Malley BW, Xu J. AIB1/SRC-3 deficiency affects insulin-like growth factor I signaling pathway and suppresses v-Ha-ras-induced breast cancer initiation and progression in mice. Cancer Res 2004;64:1875-85.
- Kuang SQ, Liao L, Wang S, Medina D, O’Malley BW, Xu J. Mice lacking the amplified in breast cancer 1/steroid receptor coactivator-3 are resistant to chemical carcinogen-induced mammary tumorigenesis. Cancer Res 2005;65:7993-8002.
- Azorsa DO, Cunliffe HE, Meltzer PS. Association of steroid receptor coactivator AIB1 with estrogen receptor-alpha in breast cancer cells. Breast Cancer Res Treat 2001;70:89-101.
- List HJ, Lauritsen KJ, Reiter R, Powers C, Wellstein A, Riegel AT. Ribozyme targeting demonstrates that the nuclear receptor coactivator AIB1 is a rate-limiting factor for estrogen-dependent growth of human MCF-7 breast cancer cells. J Biol Chem 2001;276:23763-8.
- Shao W, Keeton EK, McDonnell DP, Brown M. Coactivator AIB1 links estrogen receptor transcriptional activity and stability. Proc Natl Acad Sci USA 2004;101:11599-604.
- Planas-Silva MD, Shang Y, Donaher JL, Brown M, Weinberg RA. AIB1 enhances estrogen-dependent induction of cyclin D1 expression. Cancer Res 2001;61:3858-62.
- Reiter R, Oh AS, Wellstein A, Riegel AT. Impact of the nuclear receptor coactivator AIB1 isoform AIB1-Delta3 on estrogenic ligands with different intrinsic activity. Oncogene 2004;23:403-9.
- Harbour JW, Dean DC. The Rb/E2F pathway: expanding roles and emerging paradigms. Genes Dev 2000;14:2393-409.
- Dimova DK, Dyson NJ. The E2F transcriptional network: old acquaintances with new faces. Oncogene 2005;24:2810-26.
- Hunt KK, Keyomarsi K. Cyclin E as a prognostic and predictive marker in breast cancer. Semin Cancer Biol 2005;15:319-26.
- Keyomarsi K, Tucker SL, Buchholz TA, Callister M, Ding Y, Hortobagyi GN, et al. Cyclin E and survival in patients with breast cancer. N Engl J Med 2002;347:1566-75.
- Han S, Park K, Bae BN, Kim KH, Kim HJ, Kim YD, et al. E2F1 expression is related with the poor survival of lymph node-positive breast cancer patients treated with fluorouracil, doxorubicin and cyclophosphamide. Breast Cancer Res Treat 2003;82:11-6.
- Dhillon NK, Mudryj M. Ectopic expression of cyclin E in estrogen responsive cells abrogates antiestrogen mediated growth arrest. Oncogene 2002;21:4626-34.
- Grimberg A. Mechanisms by which IGF-I may promote cancer. Cancer Biol Ther 2003;2:630-5.
- Oh A, List HJ, Reiter R, Mani A, Zhang Y, Gehan E, et al. The nuclear receptor coactivator AIB1 mediates insulin-like growth factor I-induced phenotypic changes in human breast cancer cells. Cancer Res 2004;64:8299-308.
- Zhou G, Hashimoto Y, Kwak I, Tsai SY, Tsai MJ. Role of the steroid receptor coactivator SRC-3 in cell growth. Mol Cell Biol 2003;23:7742-55.
- Lin A, Karin M. NF-κB in cancer: a marked target. Semin Cancer Biol 2003;13:107-14.
- Werbajh S, Nojek I, Lanz R, Costas MA. RAC-3 is an NF-kappa B coactivator. FEBS Lett 2000;485:195-9.
- Wu RC, Qin J, Hashimoto Y, Wong J, Xu J, Tsai SY, et al. Regulation of SRC-3 (pCIP/ACTR/AIB-1/RAC-3/TRAM-1) coactivator activity by I kappa B kinase. Mol Cell Biol 2002;22:3549-61.
- Pages F, Vives V, Sautes-Fridman C, Fossiez F, Berger A, Cugnenc PH, et al. Control of tumor development by intratumoral cytokines. Immunol Lett 1999;68:135-9.
- Giri D, Ozen M, Ittmann M. Interleukin-6 is an autocrine growth factor in human prostate cancer. Am J Pathol 2001;159:2159-65.
- Shariat SF, Andrews B, Kattan MW, Kim J, Wheeler TM, Slawin KM. Plasma levels of interleukin-6 and its soluble receptor are associated with prostate cancer progression and metastasis. Urology 2001;58:1008-15.
- Wu RC, Qin J, Yi P, Wong J, Tsai SY, Tsai MJ, et al. Selective phosphorylations of the SRC-3/AIB1 coactivator integrate genomic responses to multiple cellular signaling pathways. Mol Cell 2004;15:937-49.
- Wu RC, Smith CL, O’Malley BW. Transcriptional regulation by steroid receptor coactivator phosphorylation. Endocrine Rev 2005;26:393-9.
- Raught B, Liao WSL, Rosen JM. Developmentally and hormonally regulated CCAAT/enhancer-binding protein isoforms influence beta-casein gene expression. Mol Endocrinol 1995;9:1223-32.
- Kagan BL, Henke RT, Cabal-Manzano R, Stoica GE, Nguyen Q, Wellstein A, et al. Complex regulation of the fibroblast growth factor-binding protein in MDA- MB-468 breast cancer cells by CCAAT/enhancer-binding protein beta. Cancer Res 2003;63:1696-705.
- Font de Mora J, Brown M. AIB1 is a conduit for kinase-mediated growth factor signaling to the estrogen receptor. Mol Cell Biol 2000;20:5041-7.
- Goel A, Janknecht R. Concerted activation of ETS protein ER81 by p160 coactivators, the acetyltransferase p300 and the receptor tyrosine kinase HER2/Neu. J Biol Chem 2004;279:14909-16.