
Metabolism and metabolic inhibition of gambogic acid in rat liver microsomes1
Introduction
Gambogic acid (GA) is a natural product isolated from the gamboge resin of Garcinia hanburyi tree in South-east Asia. The resin is used as a traditional medicine and as a color material for painting[1]. Gamboge can be used for detoxification, hemostasis, and as a parasiticide[2]. GA has been previously reported to have potent cytotoxicities against various cancer cell lines and to be a potent apoptosis inducer.
In one study, anti-tumor activity of general gambogic acids (GGA) in the experimental transplantation tumor SMMC-7721 was evaluated by relative tumor growth ratio. GGA had an inhibitory effect on the growth of SMMC-7721, which might be related to its inhibition of telomerase activity[3]. In another study, GA could inhibit the growth of SPC-A1 cells and its tumor xenografts, and when treated with GA for a period of time, telomerase activity and expression of hTERT mRNA in the tumor cells were both inhibited significantly. It is safe, at least in part, to conclude that the downregulating telomerase activity of GA by partly modifying the expression of hTERT mRNA in SPC-A1 cells may be one possible mechanism for the inhibitory activity of GA in the cells[4]. Thus GA is an effective telomerase inhibitor and displays potent anticancer activity both in vitro and in vivo. Moreover, GA was found to induce apoptosis independent of the cell cycle, which is different from paclitaxel that arrests cells in the G2/M phase[1]. The selective induction of apoptosis of GA on MGC-803 cells were studied and activation of the bax gene and suppression of the bc1-2 gene may contribute to the apoptosis mechanism[5]. Another study suggested that GA binding to transferrin receptor induces a unique signal leading to rapid apoptosis of tumor cells[6].
There are several studies about its pharmacodynamic characteristics, while only few comment on the pharmaco-kinetics. The purpose of these experiments was to investigate the metabolism of GA in rat liver microsomes in vitro and to identify the major CYP450 isoform involved in the metabolism of GA and the inhibition type of selective CYP450 inhibitor on its metabolism.
Materials and methods
Chemicals Gambogic acid was provided by the Department of Pharmaceutical Chemistry, China Pharmaceutical University. Glucose-6-phosphate dehydrogenase (Type V), α-naphthoflavone (α-Naph), sulfaphenazole (Sul), quinidine (Qui), and diethyl dithiocarbamate (DDC) were purchased from Sigma Chemical (St Louis, USA). Ketoconazole (Ket) was kindly provided by Nanjing Second Pharmaceutical Factory (Nanjing, China). α-Nicotinamide adenine dinucleotide phosphate (NADP) and glucose-6-phosphate(G-6-P) were purchased from Shanghai Lizhudongfeng Biotechnological. (Shanghai, China). All other supplies were of the highest grades available from standard commercial sources.
Tissue samples and preparation of liver microsomes Sprague-Dawley rats (180–220 g; 6–8 weeks of age) were obtained from the experimental animal center of China Pharmaceutical University, and the studies were approved by the Animal Ethics Committee of China Pharmaceutical University. The rat liver microsomes were prepared by differential centrifugation[7]. Microsomal protein concentration was determined by the method of Lowry et al[8].
Incubation and sample preparation The incubation conditions of the experiment were established and controlled to provide a reproducible and linear rate of the metabolite. A typical incubation mixture consisted of potassium phosphate buffer (PH 7.4) 100 mmol/L, an NADPH generating system (MgCl2 5 mmol/L, G-6-P 10 mmol/L, NADP 1 mmol/L, G-6-PDH 1 kU/L), gambogic acid 40 µmol/L, and microsomal protein 1 g/L, in a final volume of 1 mL[7]. The reaction was initiated by addition of the NADPH generating system. After incubation at 37 °C for 30 min, the reaction was terminated by adding 200 µL of HCl 1 mol/L. GA and its metabolites (M1 and M2) in the incubation mixture were determined by HPLC method[9] with felodipine as its internal standard. The reaction mixtures were extracted with 5 mL of ether and centrifuged at 1000×g for 10 min. The organic fraction was evaporated under a gentle stream of air at 45 °C. The residue was dissolved in 100 µL of mobile phase and an aliquot (20 µL) was injected onto the Lichrospher C18 column (250 mm×4.6 mm ID, 5 µm, Hanbon, China). These procedures were performed using a Shimadzu HPLC-UV system (Shimadzu Company, Kyoto, Japan) consisting of a LC-10ADVP pump, a SPD-10ADVP UV-VIS detector and a CTO-10A column oven. GA and its metabolites in the incubation mixture were then determined by mass analysis[9].
Inhibition study The effects of various selective CYP inhibitors on the formation of metabolite M1 in rat liver microsomes were investigated. The inhibitors studied were α-Naph (CYP1A2), Qui (CYP2D1), DDC (CYP2E1), Sul (CYP2C), and Ket (CYP3A). The concentration of GA was 40 µmol/L and the concentration range of inhibitors was 2.5–20 µmol/L for Qui, 12.5–100 µmol/L for α-Naph and Sul, 6.25–100 µmol/L for DDC, and 0.5–5 µmol/L for Ket.
Study of inhibition type After identifying the major metabolic enzyme of GA in vitro, we incubated GA in rat liver microsomes in the absence and presence of α-Naph. Then
Results
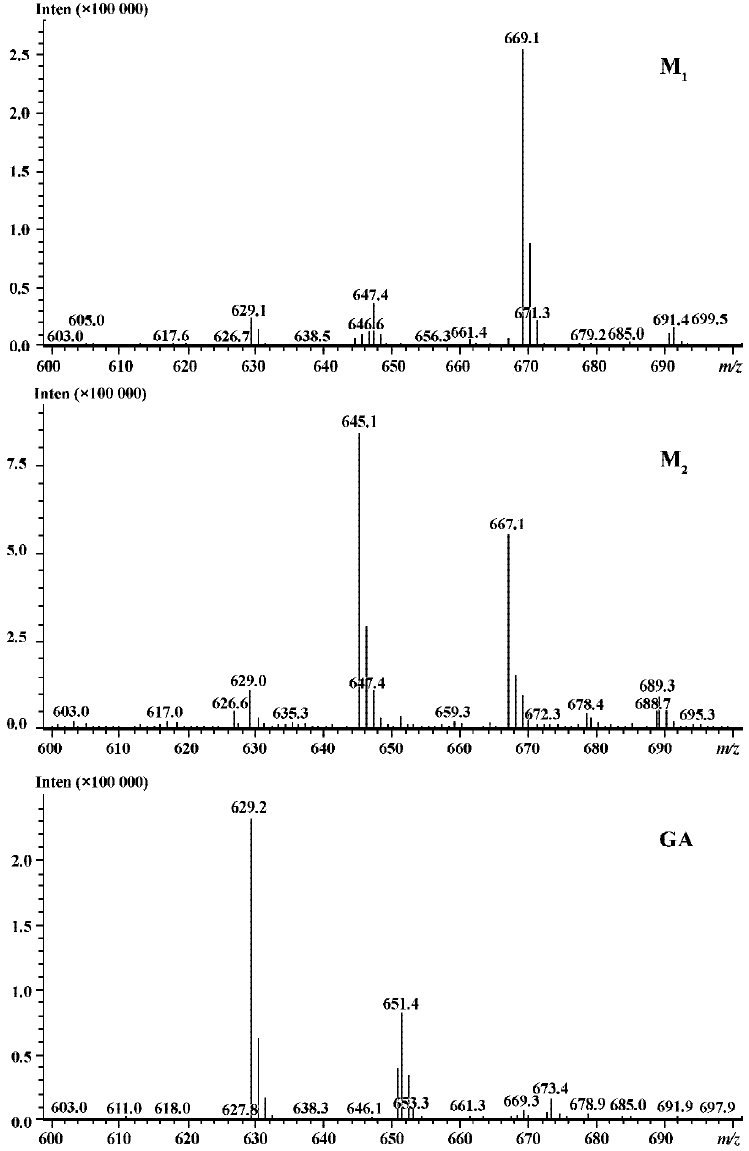
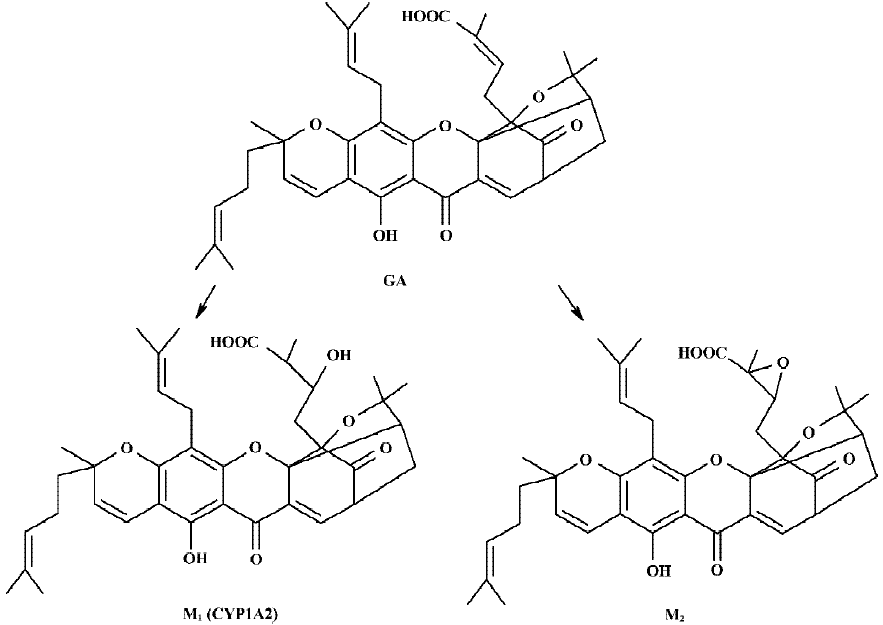
Following incubation of gambogic acid with rat liver microsomes, GA was rapidly metabolized and two metabolites (M1 and M2) were isolated in the incubation with the corresponding concentration of GA decreased (Figure 1). Direct HPLC/MS analysis[9] using an electro-spray ionization interface under positive ion mode resolved two drug-related compounds in the incubations (Figure 2). M1 exhibited the protonated molecular ion [M+Na]+ at m/z 669, [M+H]+ at m/z 647, and M2 exhibited the protonated molecular ion [M+H] + at m/z 645, [M+Na]+ at m/z 667. Thus, the molecular weight of M1 is 646 and M2 is 644. According to HPLC and mass spectra analysis, we can tentatively presume that M1 was the hydration metabolite and M2 was the epoxide metabolite of GA. But this needs further identification. The proposed metabolic pathway of GA is present in Figure 3.
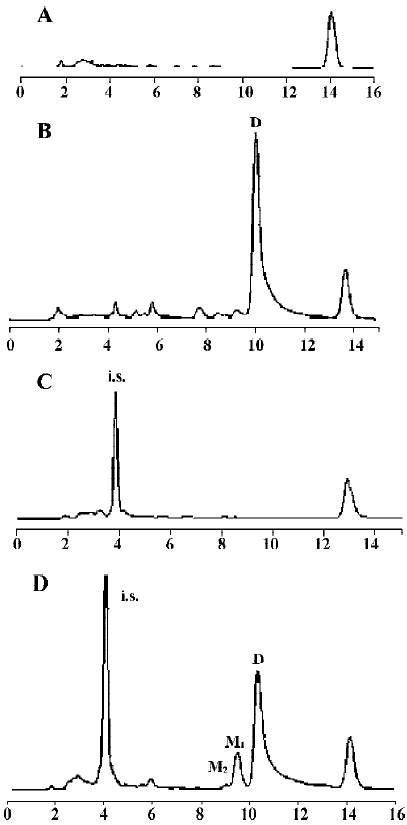
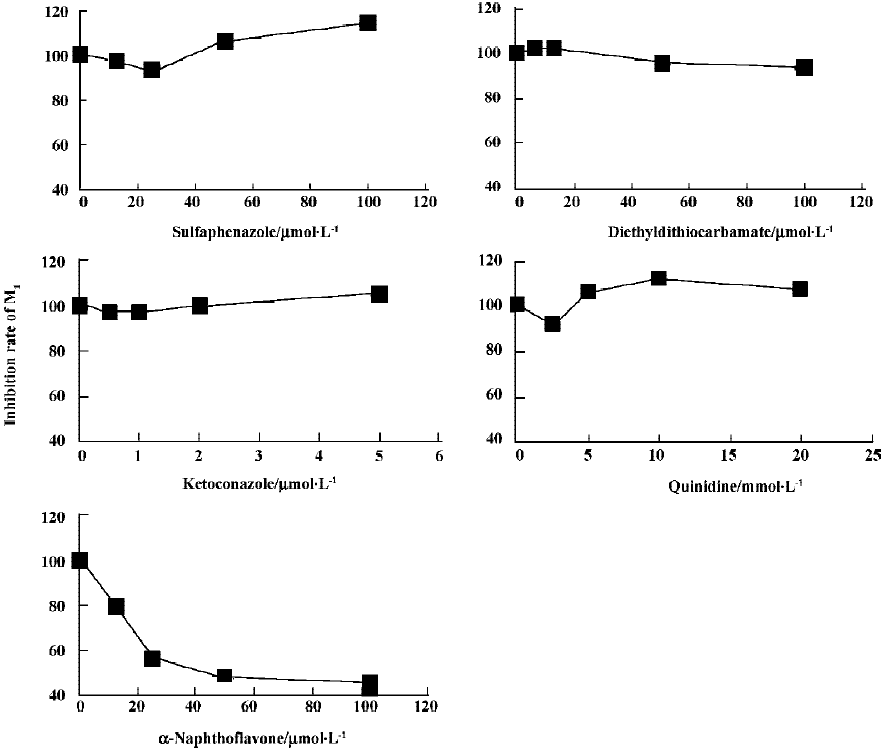
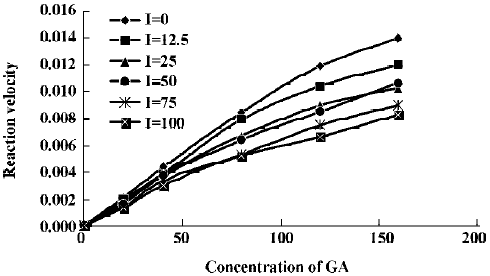
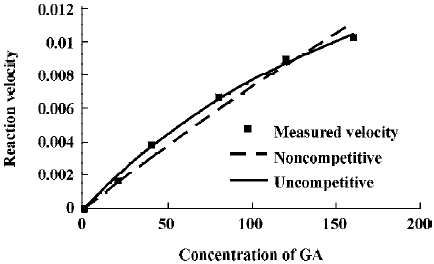
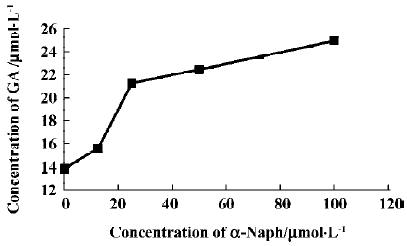
The effects of inhibitors on the formation of M1 are presented in Figure 4. α-Naph, the specific inhibitor of CYP1A2, could inhibit the formation of M1 while other inhibitors had no significant inhibitory effects on the M1 formation (Figure 3). The Michaelis-Menten plot of metabolite M1 in rat liver microsomes in the absence and presence of α-Naph is presented in Figure 5. The inhibition is parallel to the concentrations of both inhibitor and substrate, suggesting that the inhibition conforms to noncompetitive or uncompetitive types (Figure 5). Two types of inhibition in an enzyme kinetics model were then used to model the interaction: (1) the noncompetitive type, and (2) the uncompetitive type (Figure 6). As shown in Figure 6, α-Naph uncompetitively inhibited the formation of M1, resulting in a lowered rate of GA metabolism in rat liver microsomes (Figure 7).
Discussion
The results of present studies indicated that GA was rapidly metabolized in rat liver microsomes. Two metabolites were isolated and identified in the incubation mixture. In the control experiments without NADPH, only microsome and GA, there are no metabolites formed in the incubation. This suggests that the formation of M1 is NADPH-dependent. M1 was one of the major metabolites in the incubation and it was proposed to be the hydration metabolite of GA. Inhibition of the formation of this metabolite would directly result in lowing of the metabolic rate of GA. This suggested that the formation of M1 is crucial for the elimination of GA. M2 was proposed to be the epoxide metabolite of GA. Our previous studies[8] have shown that GA could be metabolized to four metabolites in rat bile in vivo, including two phase I metabolites (M1 and M2) and two phase II metabolites (M3 and M4), which were presumed to be the phase II metabolites of M1 and M2, respectively. The HPLC and LC-ESI-MS chromatographic behaviors of M1 and M2 in vitro studies showed good correlations with in vivo results.
α-Naph could uncompetitively inhibit the formation of M1 and lower the metabolism of GA, while other inhibitors for CYP3A (Ket), CYP2C (Sul), CYP2D1 (Qui) and CYP2E1 (DDC) had little or no effect on the formation of M1. The results suggest that CYP1A2 is a major CYP450 isoform involved in the metabolism of GA. Pharmacokinetic drug interactions caused by metabolic processes are regarded as one of the most important factors affecting the concentration of drugs[10]. Therefore, inhibition of the above metabolic pathway can lower the formation rate of M1 and increase the concentration of GA, suggesting the possibilities of metabolic interactions of GA with other therapeutic agents, such as inhibitors and substrates of CYP1A2. In clinical applications, drug-drug interactions often occur when two or more drugs are used in combination, which can result in severe side effects in some cases[11]. This is worth noting when GA is used in combination with other therapeutic agents that may have metabolic interactions with it. All of the above results contribute to our understanding of the metabolism of GA in rats.
Reference
- Zhang HZ, Kasibhatla S, Wang Y, Herich J, Guastella J, Tseng B, et al. Discovery, characterization and SAR of gambogic acid as a potent apoptosis inducer by a HTS assay . Bioorg Med Chem 2004;12:309-17.
- Liu W, Guo QL, Li Z, Gu HY. Anticancer effect and apoptosis induction of gambogic acid in human gastric cancer line BGC-823. World J Gastroenterol 2005;11:3655-9.
- Guo QL, You QD, Wu ZQ, Yuan ST, Zhao L. General gambogic acids inhibited growth of human hepatoma SMMC-7721 cells in vitro and in nude mice. Acta Pharmacol Sin 2004;25:769-74.
- Wu ZQ, Guo QL, You QD, Zhao L, Gu HY. Gambogic acid inhibits proliferation of human lung carcinoma SPC-A1 cells in vivo and in vitro and represses telomerase activity and telomerase reverse transcriptase mRNA expression in the cells. Biol Pharm Bull 2004;27:1769-74.
- Zhao L, Guo QL, You QD, Wu ZQ, Gu HY. Gambogic acid induces apoptosis and regulates expression of Bax and Bcl-2 protein in human gastric carcinoma MGC-803 cells. Biol Pharm Bull 2004;27:998-1003.
- Kasibnatla S, Jessen KA, Maliartchouk S, Wang JY, English NM, Drewe J, et al. A role for transferrin receptor in triggering apoptosis when targeted with gambogic acid. Cell Biol 2005;102:12095-100.
- Liu XQ, Zhao Y, Li D, Qian ZY, Wang GJ. Metabolism and metabolic inhibition of clinipine in human liver microsomes. Acta Pharmacol Sin 2003;24:263-8.
- Dawson JM, Heatlie PL. Lowry method of protein quantification: evidence for photosensitivity. Anal Biochem 1984;140:391-3.
- Hao K, Liu XQ, Wang GJ. Pharmacokinetics of gambogic Acid in rats. J China Pharm Univ 2005;36:338-41. Chinese.
- Nakajima M, Inoue T, Shimada N, Tokudome S, Yamamoto T, Kuroiwa Y. Cytochrome P4502C9 catalyzes indomethacin O-demethylation in human liver microsomes. Drug Metab Dispos 1998;26:261-6.
- Bjornsson T, Callaghan J, Einolf H, Fischer V, Gan L, Grimm S, et al. The conduct of in vitro and in vivo drug-drug interaction studies: A Pharmaceutical Research and Manufacturers of America (PhRMA) perspective. Drug Metab Dispos 2003;31:815-32.