
FAK-related nonkinase attenuates hypertrophy induced by angiotensin-II in cultured neonatal rat cardiac myocytes
Introduction
Cardiac hypertrophy is an adaptive response of the heart to a variety of intrinsic and extrinsic stimuli, including hypertension, myocardial infarction, endocrine disorders, valvular diseases and so on. Although hypertrophy is initially beneficial, prolonged hypertrophy can ultimately become deleterious and result in cardiomyopathy, heart failure, and sudden death[1]. Elucidation of the molecular mechanisms that activate and maintain the hypertrophic program represents a major challenge. Previous studies have demonstrated that activation of focal adhesion kinase (FAK) has been implicated in the progress of cardiomyocyte hypertrophy. FAK-related nonkinase (FRNK) is an alternate transcript of the C-terminal region of the FAK gene and acts as an endogenous regulator of FAK activity[2]. However, how FRNK inhibit angiotensin-II-induced cardiac hypertrophy is still not well understood. In the present study, we investigated the effect of FRNK on cardiomyocyte hypertrophy induced by angiotensin-II, and focused on the changes of relative signal molecules to reveal the possible anti-hypertrophy mechanisms of FRNK.
Materials and methods
Materials Neonatal Sprague-Dawley rats were purchased from the animal supply center of Tongji Medical College. TRIzol reagent was purchased from Life Technologies, Inc (Gaithersburg, MD, USA) and the RT-PCR kit was from TaKaRa (Dalian, China). Restriction enzymes and DNA Marker were from MBI Fermentas (Hanover, MD, USA). DMEM culture medium and fetal bovine serum were purchased from GIBCO (Grand Island, NY, USA). Enhanced chemiluminescence reagent was from Pierce Chemical (Rockford, IL, USA), prestained molecular weight standards were from Bio-Rad, Inc (Hercules, CA, USA), PVDF membranes were from Schleicher and Schuell (Dassel, Germany) and rabbit polyclonal (C-20) antibody against the C-terminus of focal adhesion kinase, antibodies against ERK1/2, p-ERK1/2, NF-κB RelA/p65 and β-actin were all purchased from Santa Cruz Biotechnology Inc (Santa Cruz, CA, USA). Antibodies against AKT and p-AKT were purchased from Cell Signaling Technology, Inc (Beverly, MA, USA).
Preparation of pcDNA3.1-FRNK
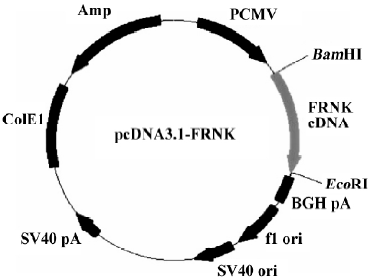
Construction of pcDNA3.1-FRNK Total RNA was extracted from fresh intestine tissue isolated from 18-day chick embryogenesis (where FRNK expresses at a high level[3]). Upstream primer (5'-GTT GGA TCC GTC AGA ACT GTG TAC AAT ACT GGA GGA GG-3') and downstream primer (5'-CTC GAA TTC CAA CAG ACA GAT GAA ACC TTC CCT C-3') were used to amplify chick FRNK gene and restriction endonucleases of BamHI and EcoRI were boldfaced, respec-tively. According to the manufacturer’s instructions for the RT-PCR kit, the first-strand cDNA synthesis was carried out in a 20 µL reaction volume with 1 µg of total RNA and the following components: 1 µL (2.5 pmol/mL) of Oligo (dT), 1 μL total RNA (1 µg/µL), 8.5 μL of RNA-free double distilled H2O, 2 µL of 10×RNA PCR buffer [250 mmol/L Tris-HCl (pH 8.3), 375 mmol/L KCl, and 15 mmol/L MgCl2], 2 µL of a 10 mmol/L dNTP mix, 0.5 µL of RNase inhibitor (40 U/µL), and 1 µL of avian myeloblastosis virus (AMV) reverse transcriptase (5 U/µL). The mixture was incubated at 42 °C for 30 min, and then at 99 °C for 5 min. The PCR reaction was performed in a final reaction volume of 25 μL using 5 μL of the reverse transcription reaction product in a thermal cycle with a 1-min denaturing phase at 95 °C, a 45-s annealing phase at 55 °C, and a 3-min extension phase at 72 °C for 35 cycles. The amplified products were analyzed with 1% agarose gel electrophoresis (Figure 1), and recovered with a Qiaquick gel extraction kit. The RT-PCR products were then digested by BamHI and EcoRI and was ligated with pcDNA3.1 vector at the site digested by the same restriction endonucleases in the presence of T4 DNA ligase at 16 °C overnight, forming a new plasmid pcDNA3.1-FRNK (Figure 2). The ligation mixtures were transformed into Ecoli DH5α, and then spread on an LB agarose plate containing ampicillin (100 µg/mL). The correct plasmid of recombinant FRNK eukaryotic expression vector identified by the restriction analysis was further confirmed using an automated DNA sequencer (TaKaRa) and was subsequently compared with the published gallinaceous cDNA sequence.
Plasmid purification by cesium chloride density gradient centrifugation A colony of bacteria harboring the recombinant FRNK were put into an Ehrlenmeyer flask containing 1 L of TB media supplemented with ampicillin at 100 μg/mL and incubated at 37 °C for 12–16 h with shaking at 750×g. The cells were harvested by centrifugation at 24 000×g for 20 min in 500 mL bottles and plasmid DNA was recovered from bacterial cells by an alkaline lysis procedure. The closed circular plasmid DNA was then purificated by CsCl/EB as described in a previous study[4]. The concentration of the DNA was measured by the spectrophotometer and plasmid was stored at -20 °C.
Cardiomyocyte culture and treatments Neonatal cardiomyocytes were isolated and cultured according to the method described by Simpson and Savion[5], with some modifications. The ventricles of neonatal SD rats (1 to 3 d old) were digested with 0.1% pancreatin at 37 °C. Dissociated cells were preplated for 1 h in serum-free DMEM medium to selectively remove nonmuscle cells. Myocytes were then plated in 6-well dishes (1×106 cells/well) in a 5% CO2 incubator for 18 h. Unattached cells were removed by aspiration, and the attached cells were maintained in DMEM solution containing 20% fetal bovine serum for an additional 48 h. The medium was then changed to serum-free cultured DMEM solution. The next day, myocytes were divided into four groups. Two groups of cells were respectively transfected with pcDNA3.1-FRNK (FRNK group) or pcDNA3.1 (pcDNA3.1 group) by Lipofectamine 2000 according to the manufacturer’s instructions. At 24 h after transfection, the two groups were treated with angiotensin-II (1×10-6 mol/L) for a further 48 h. The Ang II group was treated only with angiotensin-II (1×10-6 mol/L) at the same time. The blank group that had received no treatment served as control.
Measurement of surface area of cardiac myocytes The surface area of cardiac myocytes was measured according to the protocol of Simpson[6]. Surface area was quantified by imaging to the complete boundary of 30 individual cells/group using image analysis software (SCION IMAGING; National Institutes of Health, USA). From this, a mean±SEM value was calculated for the surface area in each group.
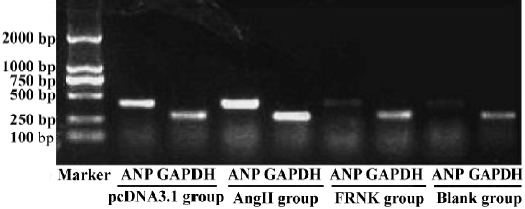
RT-PCR analysis of ANP mRNA Total RNA was extracted from myocytes with TRIzol reagent. Analysis of the expression of ANP mRNA was done using RT-PCR technique. Expression of mRNA for glyceraldehydes-3-phosphate dehydrogenase (GAPDH) was used as an internal standard. Total RNA (0.5 µg) was reverse transcribed in a final volume of 20 µL with AMV reverse transcriptase (5 U/µL) and 1 µL oligo (dT) primer (2.5 pmol/mL) according to the RT-PCR manufacturer’s protocol. Single-strand cDNA template (5 L) was used for polymerase chain reaction (PCR) to amplify a 410-bp fragment for ANP with the complementary antisense primer (5'-GGC TCC TTC TCC ATC ACC AA-3') and the sense primer (5'-CCA ATC CTG TCA ATC CTA CC-3'). For a 274-bp fragment for the housekeeping gene GAPDH, the antisense primer was 5'-GCT CAT GAC CAC AGT CCA-3', the sense primer 5'-CTG CTT CAC CAC CTT CTT-3'. The conditions for PCR were one cycle of denaturation at 95 °C for 5 min followed by 30 cycles of 95 °C for 60 s, 56 °C for 45 s, and 72 °C for 60 s, with a final extension at 72 °C for 7 min. The electrophoresed PCR products were scanned by densitometry and the ratio of ANP to GAPDH was calculated in each sample.
Total protein, nuclear extracts and Western blot
Protein isolation Myocytes were incubated with the Tris buffer on ice for 30 min, and then removed from the 6-well plates by cell scraping. Subsequently, the lysate was centrifuged at 4 °C for 10 min and the supernatants (total protein) were collected. Nuclear extracts were isolated using a method described in a previous study[7]. The concentrations of total and nuclear proteins were quantified by the Bradford method.
Western blot Total or nuclear protein samples (20 µg per lane) were separated by 10% SDS/PAGE, electrophoretically transferred onto PVDF membranes, and the membranes were incubated for 2 h at room temperature with 5% nonfat dried milk in 10 mmol/L Tris-HCl (pH 7.5), 100 mmol/L NaCl, and 0.1% Tween-20 (TBS-T). For nuclear extracts, the membrane was incubated with a rabbit anti-NF-κB RelA/p65 antibody and, for total proteins, the membrane was incubated with appropriate dilutions of primary antibody against the C-terminus of FAK, ERK1/2, p-ERK1/2, AKT and p-AKT, followed by incubation with an appropriate dilution of the secondary antibody for 2 h at room temperature. After each incubation, the blot was washed 4 times in TBS-T at room temperature and then developed with enhanced chemilumine-scence.
Statistical analysis The data are expressed as mean±SEM and analyzed by means of either Student’s t-test or ANOVA, with the use of SPSS 12.0 software. Differences were considered statistically significant at P< 0.05.
Results
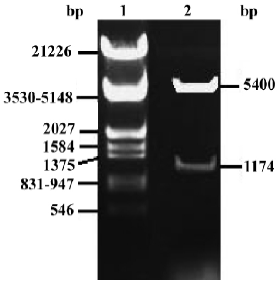
Identification of pcDNA3.1-FRNK To demonstrate whether the full-length chick FRNK-cDNA was inserted into the multi-clone sites of pcDNA3.1 correctly, pcDNA3.1-FRNK was digested by BamHI and EcoRI and evaluated with electrophoresis (Figure 3).
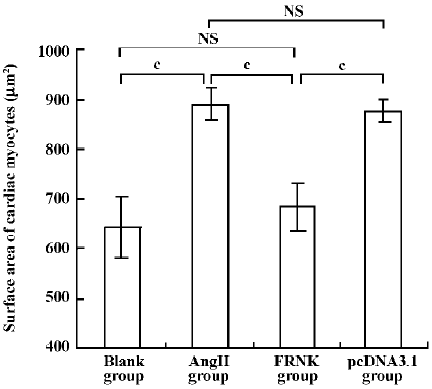
Effect of FRNK on surface area in angiotensin-II-induced hypertrophic myocytes To assess cellular hypertrophy, the surface area of variant treated cardiac myocytes was quantified (Figure 4). The average surface area of cardiac myocytes in the Ang II group increased by ~38.0% compared to the blank group (889±32 µm2 vs 644±60 µm2). However, cell surface area in the FRNK group was significantly reduced compared with the Ang II or pcDNA3.1 groups (684±47 µm2 vs 889±32 µm2, 684±47 µm2 vs 877±23 µm2; P< 0.01). These data indicate that FRNK could decrease the surface areas of hypertrophic myocytes induced by angiotensin-II.
Effect of FRNK on expression of ANP mRNA Pathological cardiac hypertrophy is usually associated with the re-expression of the fetal gene program[8]. So we examined the expression of ANP mRNA (Figure 5). Cardiac myocytes in the AngII and pcDNA3.1 groups resulted in significant increases in the levels of ANP mRNA (ODANP/GAPDH =1.13±0.06, 1.09±0.05, respectively). But the expression of ANP mRNA in the FRNK group was dramatically lower than that in the Ang-II group. We detected no significant changes in the expression of the ANP gene between myocytes with the Blank group (ODANP/GAPDH = 0.29±0.04) and FRNK group (ODANP/GAPDH=0.35±0.02). The results indicated that FRNK suppressed the re-expression of ANP mRNA in hypertrophic myocytes.
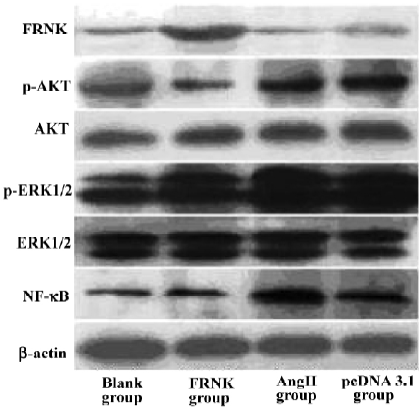
Effects of FRNK overexpression on Ang II-induced signal transduction At first, we used the C-terminal anti-FAK antibody to examine the FRNK expression in different groups. Obviously, the protein level of FRNK in the FRNK group was higher than that in other groups. We then examined the expression of intracellular signal molecules because many aspects of cardiac myocyte growth and function are dependent on signal pathways. We found no evident effect of expressions of total ERK1/2 and AKT in each group. However, strikingly higher expressions of ERK1/2 and AKT phosphorylation in the Ang II group were seen in contrast to the Blank group. In the FRNK group, markedly attenuated ERK1/2 and AKT phosphorylation was observed, but in the pcDNA3.1 group there were the same levels of the expression compared to Ang II group. Subsequently, we also examined the NF-κB p65 protein level in the nucleus. Immunoblotting of nuclear extracts with an anti-NF-κB p65 antibody showed low expressions of NF-κB p65 protein in the FRNK and Blank groups, while stronger expression was observed in the Ang II and pcDNA3.1 groups (Figure 6). This suggested that FRNK overexpression regulated hypertrophic signaling through decreasing ERK1/2 and AKT phosphorylation, and subsequently downregulation of NF-κB p65 in the nucleus.
Discussion
Activation of FAK has been implicated in the progress of cardiomyocyte hypertrophy[9]. Focal adhesion formation triggers FAK autophosphorylation of Tyr397 and activation, leading to the recruitment of SH2 and SH3 domain-containing kinases such as Src and Fyn and subsequent phosphorylation of other cytoplasmic plaque proteins such as paxillin, tensin, and p130CAS. Direct Grb2 or Shc binding to FAK and the indirect association of the SOS GDP-GTP exchange factor for Ras with Grb2 can link FAK to the activation of the Ras-ERK2/MAP kinase pathway[10]. FAK also influences other signaling molecules such as PI-3K/AKT, PLC, PKCα, PKCδ and PKCε[11]. Thus, FAK may function as a “scaffold” for the assembly of signaling complexes through the coordinated recruitment of other signaling and adaptor proteins[12]. FRNK is the autonomously expressed, noncatalytic C-terminal portion of FAK. Because FRNK does not possess catalytic activity but comprises the focal adhesion target domain, FRNK can compete with FAK for focal adhesion binding sites and acts as an endogenous regulator of FAK activity[2,13].
Ang II has been implicated in cardiac hypertrophy[14]. In the present study, cardiac myocytes incubated with angiotensin-II (1×10-6 mol/L) for 48 h, resulting in a significant increase of surface area of myocytes, while the surface area of myocytes transfected with pcDNA3.1-FRNK in advance was reduced. Because it has long been recognized that the upregulation of the so-called fetal gene ANP was associated with the development of pathological cardiac hypertrophy[8], the mRNA levels of ANP in differently treated groups were also examined. Our data indicated that transfection of pcDNA3.1-FRNK indeed decreased the mRNA expression of ANP in hypertrophic myocytes.
The detailed mechanisms by which FRNK regulates hypertrophic growth of cardiomyocytes are not known. Many lines of evidence have suggested that ERK1/2 and AKT are key molecules in intracellular signal transduction and play essential roles in cellular proliferation and differentiation in many cell types, including cardiomyocytes[15,16]. In the present study, we tested the expression of ERK1/2 and AKT, as well as phosphorylation levels of ERK1/2 and AKT in different groups. Furthermore, we examined the level of NF-κB p65 in the nucleus. NF-κB is known to be involved in inflammation, the immune response, viral infection, and the regulation of programmed cell death[17]. Recent studies by several investigators have also implicated NF-κB activation as a causal event in cardiac hypertrophic response[18,19]. As we know, the NF-κB family has 5 mammalian members: p50, p52, c-Rel, p65, and RelB. In most resting cells, NF-κB is a p50/p65 dimer that is retained in the cytoplasm. On stimulation, the NF-κB p65 subunit is translocated into the nucleus and regulates gene transcription[20]. In this report, we showed that the overexpression of FRNK in myocytes led to decreasing levels of ERK1/2 and AKT phosphorylation, and NF-κB p65 protein in nuclear extracts, but did not influence total expressions of ERK1/2 and AKT. These results, together with previously published data[10–11,15,16], indicate that FRNK inhibits angiotensin-II-induced cardiomyocyte hypertrophy by affecting Raf-1 kinase/ERK and PI-3K/AKT signal pathways, and then down-regulation of nuclear translocation of NF-κB p65.
In summary, our study demonstrates that overexpression of FRNK has beneficial effects on the hypertrophic myocytes stimulated by angiotensin-II. FRNK may be a therapeutic target for the treatment of cardiac hypertrophy.
References
- Pokharel S, Sharma UC, Pinto YM. Left ventricular hypertrophy: virtuous intentions, malignant consequences. Int Biochem Cell Biol 2003;35:802-6.
- Richardson A, Parsons JT. A mechanism for regulation of the adhesion-associated protein tyrosine kinase pp125FAK. Nature 1996;380:538-40.
- Nolan K, Lacoste J, Parsons JT. Regulated expression of focal adhesion kinase-related nonkinase, the autonomously expressed C-terminal domain of focal adhesion kinase. Mol Cell Biol 1999;19:6120-9.
- Good L, Nazar RN. Visualization of CsCl/EtdBr plasmid preparations under visible light. Biotechniques 1995;18:556-8.
- Simpson P, Savion S. Differentiation of rat myocytes in single cell culture with and without proliferating nonmyocardial cells. Circ Res 1982;50:101-16.
- Simpson P. Stimulation of hypertrophy of cultured neonatal rat heart cells through an alpha 1-adrenergic receptor and induction of beating through an alpha 1- and beta 1-adernergic receptor interaction. Circ Res 1985;56:884-94.
- Schreiber E, Matthias P, Muller MM, Schaffner W. Rapid detection of octamer binding proteins with “mini-extracts”, prepared from a small number of cells. Nucleic Acids Res 1989;17:6419.
- Harada M, Itoh H, Nakagawa O, Ogawa Y, Miuamoto Y, Kuwahara K, et al. Significance of ventricular myocytes and nonmyocytes interaction during cardiocyte hypertrophy: evidence for endothelin-1 as a paracrine hypertrophic factor from cardiac nonmyocytes. Circulation 1997;96:3737-44.
- Sharp WW, Simpson DG, Borg TK, Samarel AM, Terracio L. Mechanical forces regulate focal adhesion and costamere assembly in cardiac myocytes. Am J Physiol 1997;273:H546-56.
- Shen TL, Guan JL. Differential regulation of cell migration and cell cycle progression by FAK complexes with Src, PI3K, Grb7 and Grb2 in focal contacts. FEBS Lett 2001;499:176-81.
- Reif S, Lang A, Lindquist JN, Yata Y, Gabele E, Scanga A, et al. The role of focal adhesion kinase-phosphatidylinositol 3-kinase-akt signaling in hepatic stellate cell proliferation and type I collagen expression. J Biol Chem 2003;278:8083-90.
- Schlaepfer DD, Hauck CR, Sieg DJ. Signaling through focal adhesion kinase. Prog Biophys Mol Biol 1999;71:435-78.
- Hayasaka H, Simon K, Hershey ED, Masumoto KH, Parsons JT. FRNK, the autonomously expressed C-terminal region of focal adhesion kinase, is uniquely regulated in vascular smooth muscle: analysis of expression in transgenic mice. J Cell Biochem 2005;95:1248-63.
- Schultz Jel J, Witt SA, Glascock BJ, Nieman ML, Reiser PJ, Nix SL, et al. TGF-beta 1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin-II. J Clin Invest 2002;109:787-96.
- Pages G, Lenormand P, Lallemain G, Chambard JC, Meloche S, Pouyssegur J. Mitogen-activated protein kinase p42mapk and p44mapk are required for fibroblast proliferation. Proc Natl Acad Sci USA 1993;90:8319-23.
- Calautti E, Li J, Saoncella S, Brissette JL, Goetinck PE. Phosphoinositide 3-kinase signaling to Akt promotes keratinacyte differentiation versus death. J Biol Chem 2005;280:32856-65.
- Baeuerle PA, Baltimore D. NF-κB: ten years after. Cell 1996;87:13-20.
- Purcell NH, Tang G, Mercurio F, DiDonato JA, Lin A. Activation of NF-κB is required for hypertrophic growth of primary rat neonatal ventricular cardiomyocytes. Proc Natl Acad Sci USA 2001;98:6668-73.
- Gupta S, Purcell NH, Lin A, Sen S. Activation of nuclear factor-κB is necessary for myotrophin-induced cardiac hypertrophy. J Cell Biol 2002;159:1019-28.
- Gupta S, Young D, Sen S. Inhibition of NF-kappaB induces regression of cardiac hypertrophy, independent of blood pressure control, in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol 2005;289:H20-9.