
Acute pulmonary inflammation is inhibited in CXCR3 knockout mice after short-term cigarette smoke exposure1
Introduction
Cigarette smoke (CS) is the most important risk factor for CS-related pulmonary pathophysiology[1]. CS induces the release of inflammatory cytokines, such as interleukin (IL)-1 and -8, from bronchial epithelial cells through the oxidative pathway, accounting for potential neutrophil and monocytic chemotactic activities released from the epithelium[2]. The view that CS induced-inflammation plays a predominant role in chronic obstructive pulmonary disease (COPD) is further supported by findings of excessive CD8+ T cells in lung tissues from COPD patients, and increased CD8+ T cells in the epithelium and submucosa expressing chemokine C-X-C motif receptor 3 (CXCR3+) in smokers with COPD[3–5]. CXCR3, together with its specific ligand CXCL10/interferon (IFN)-inducible protein-10 (IP-10), which is abundantly expressed in bronchiolar epithelial cells and airway smooth muscle cells, has been demonstrated to play a pivotal role in mediating T-cell (particularly Th1/Tc1) recruitment into the airways and lung parenchyma upon CS exposure[5].
The role of CXCR3 in initiating acute pulmonary inflammation induced by CS has not previously been studied. In this present study, we used CXCR3 gene knockout (CXCR3–/–) mice and their wild-type (WT) counterparts to investigate the contribution of CXCR3 in an acute inflammatory event induced by CS after short-term exposure.
Materials and methods
Mice and CS exposure The CXCR3 gene-deficient mouse line has been established by gene targeting, as described elsewhere[6], and was kindly provided by Dr Bao LU (Harvard Medical School, Boston, MA, USA). The CXCR3–/– and WT mice (Experimental Animal Research Center, Beijing, China) with a C57BL/6 background (backcross>14 generations), were maintained under a pathogen-free conditions at a mouse facility at Peking Union Medical College (Beijing, China). Clean food and water were given ad libitum; 10–12-week-old mice (20–22 g) were used in the experiments.
A commercially-available cigarette was used (White Shark; Tobacco Company, China), and according to manufacturer’s specifications, each cigarette contained 1 mg nicotine and 13 mg tar. CS exposure was performed according to previously described methods[7–9]. Briefly, the whole body of mice (n=8 groups) were exposed to the mainstream of the tobacco smoke of 5 cigarettes 4 times a day with a 30 min smoke-free interval for 3 consecutive days. The control mice received filtered air according to the same procedure.
The animals were killed at 2 h after the last CS exposure by pentobarbital overdose.
All experiments were performed according to international and institutional guidelines for animal care, and approved by the Peking Union Medical College Hospital Committee on the Animal Care and Use.
Preparation of lung tissues and morphometrical evaluation of pulmonary inflammation The animals were killed, and the lungs were removed, inflated to 25 cm with H2O and 10% formalin, fixed overnight, embedded in paraffin, and sectioned at 5 µmol/L. Hematoxylin-eosin (HE) staining was performed at the Department of Pathology, Peking University Health Sciences Center (Beijing China). Histopathological assessments in the bronchus and the parenchyma were performed in a blind manner by one of the authors (Dr Wei-xun ZHOU).
Bronchoalveolar lavage The mice were killed and a 20 gauge catheter was inserted into the trachea. Bronchoalveolar lavage (BAL) was performed twice with 0.8 mL ice-cold phosphate-buffered saline (PBS; pH 7.4); 1.5 mL of total injected volume was recovered in >95% of mice. The BAL fluid was centrifuged at 1500 r/min for 5 min at 4 oC, and the supernatant was collected for the measurement of cytokines. The cells were harvested, red cells were lysed, and the cell pellets were washed and suspended in cold PBS. Total cells were enumerated by counting on a hemocytometer. For differential cell counting, cells were spun onto glass slides, fixed, and stained. The number of macrophages, neutrophils, and lymphocytes in 400 cells was counted based on morphology.
Lung homogenates The animals were anesthetized and lavaged with 3 mL cold saline via the heart. The left lobes were removed and homogenized in 1 mL PBS containing complete protease inhibitor cocktail (Sigma, St Louis, MO, USA). The samples were then centrifuged and filtered through a 0.45 μm filter and kept at –70 oC until use.
Preparation of lung single-cell suspensions The lungs were excised, minced, and digested for 30 min in 15 mL digestion buffer [RPMI-1640, 10% fetal bovine serum (FBS), 1% penicillin/streptomycin, 1 mg/mL collagenase (Sigma, USA), and 30 µg/mL DNase (Sigma, USA)]. The undigested fragment was further dispersed by repeated passage through a Nytex filter (Tetko, Kansas City, MO, USA). The total cells were harvested and suspended in 10 mL completed medium (RPMI-1640, 10% FBS, and 1% penicillin/streptomycin). An equal volume of 40% Percoll was added, and the cells were centrifuged at 3000 r/min for 30 min at room temperature. The cell pellets were resuspended in complete medium, and leukocytes were counted on a hemocytometer in the presence of 0.4% trypan blue. Cells were >90% viable by trypan blue exclusion. Cytospins of recovered cells were prepared for differential staining, as described earlier.
Labeling cells from the BAL fluid and single-cell suspensions from lung tissues In total, 50 µL of 2×107/mL cells from the BAL fluid and digested lung cells was used, and 10 µL blocking buffer (1 µL blocking antibody Fc in 9 mL PBS/2% bovine serum albumin) was added to the cells for 15 min on ice to block non-specific binding. After washing once, the cells were incubated with 50 µL fluorescein-isothiocyanate-conjugated anti-CD4 antibody and PE-conjugated anti-CD8 antibody or control mouse immunoglobulin G2b (BD Pharmingen, San Diego, CA, USA) for 1 h on ice. The cells were washed twice by PBS and fixed in PBS containing 2% formalin. The cells were subjected to flow cytometry on a FACScan cytometer (Coulter, Hialeah, FL, USA).
RNA extraction and semiquantitative RT-PCR analysis Total RNA was extracted from the lungs using TRIzol reagent according to the manufacturer’s instructions, and treated with RNase-free DNase. RNA was reverse transcribed, and cDNA was subjected to PCR for analyzing the expression of TNF-α, IFN-γ, transforming growth factor (TGF)-β1, CXCL9, CXCL10, CXCL11, and β-actin. The primers and conditions for the PCR are detailed in Table 1. All PCR reagents were purchased from Invitrogen (Carlsbad, CA, USA).
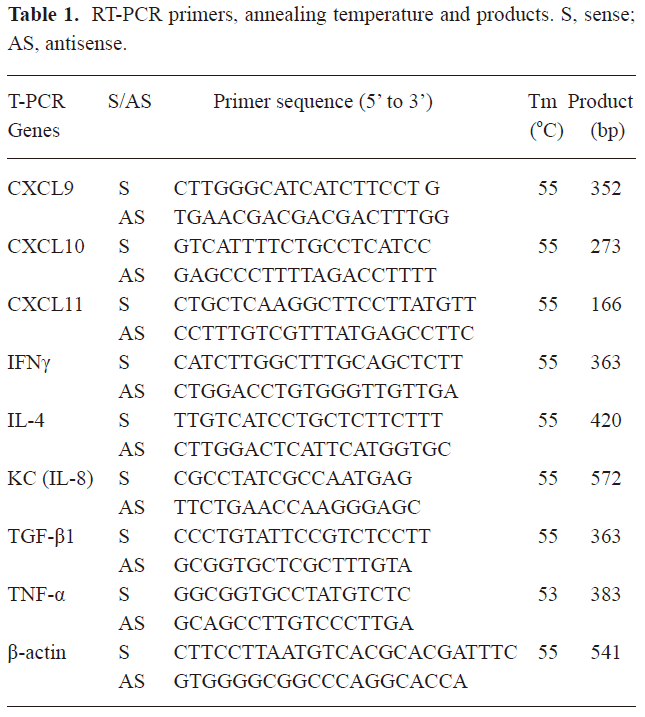
Full table
ELISA analysis of cytokines The concentrations of TNF-α, IL-4, IFN-γ, and CXCL10 in the BAL fluid and lung homogenates were determined by ELISA kits (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s recommendations.
Statistical analysis Data are expressed as mean± SEM. Comparisons between 2 groups were carried out with the non-parametric two-tailed t-test using GraphPad PRISM software (version 4.0 for Windows; GraphGrad, San Diego, CA, USA) as needed. A value of P<0.05 was considered significant.
Results
Analysis of infiltration of inflammatory cells into the airways and lungs In the murine model used in this study, 3 d of CS exposure caused a strong inflammatory response, including a significant infiltration of neutrophils, lymphocytes, and macrophages, particularly in the BAL fluid from WT mice at 2 h after 3 d of exposure to CS. By contrast, there was a mild infiltration of inflammatory cells into the airways and lung tissues in the CXCR3–/– mice compared with the WT mice (Table 2).
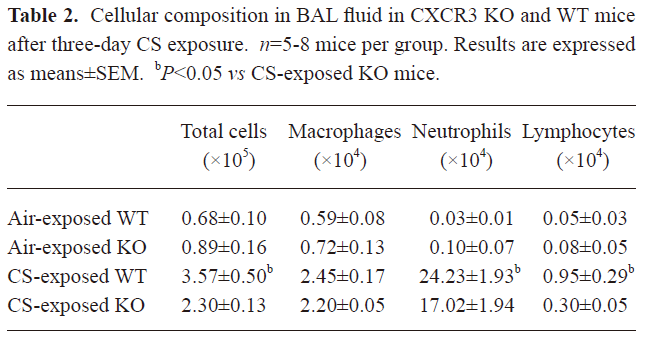
Full table
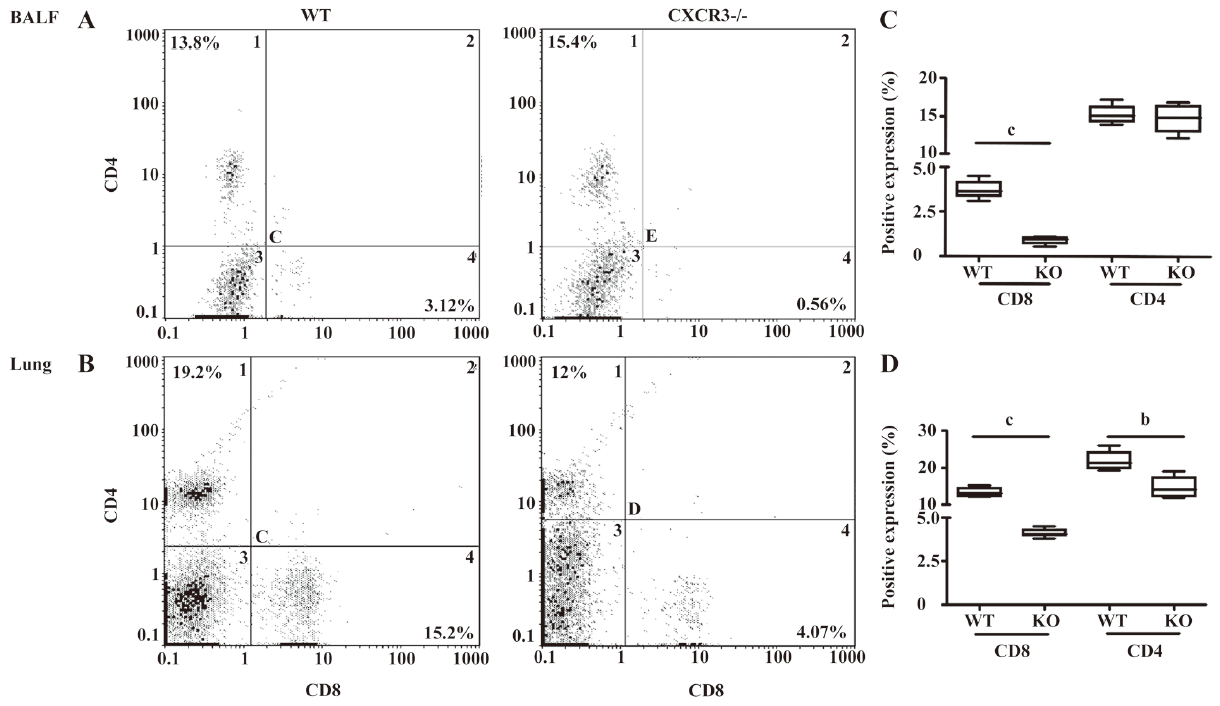
The percentage of CD8+ T cells in the BAL fluid from CXCR3–/– mice significantly decreased compared to the CXCR3 WT mice after CS exposure (0.9%±0.1% vs 3.7%±0.3%, P<0.01), while the percentage of CD4+ T cells did not significantly differ between the 2 groups (14.7%±1.1% vs 15.3%±0.7%, P=0.62; Figure 1A). The proportion of CD8+ T was profoundly decreased in the lung tissues of the CXCR3–/– mice compared to the WT mice (4.1%±0.1% vs 13.4%±0.7% for 2 h; Figure 1B). The percentage of CD4+ T cells was significantly higher in the lungs of the WT mice than in their CXCR3–/– counterparts (22.0%±1.5% vs 14.7%±1.6%, P<0.05).
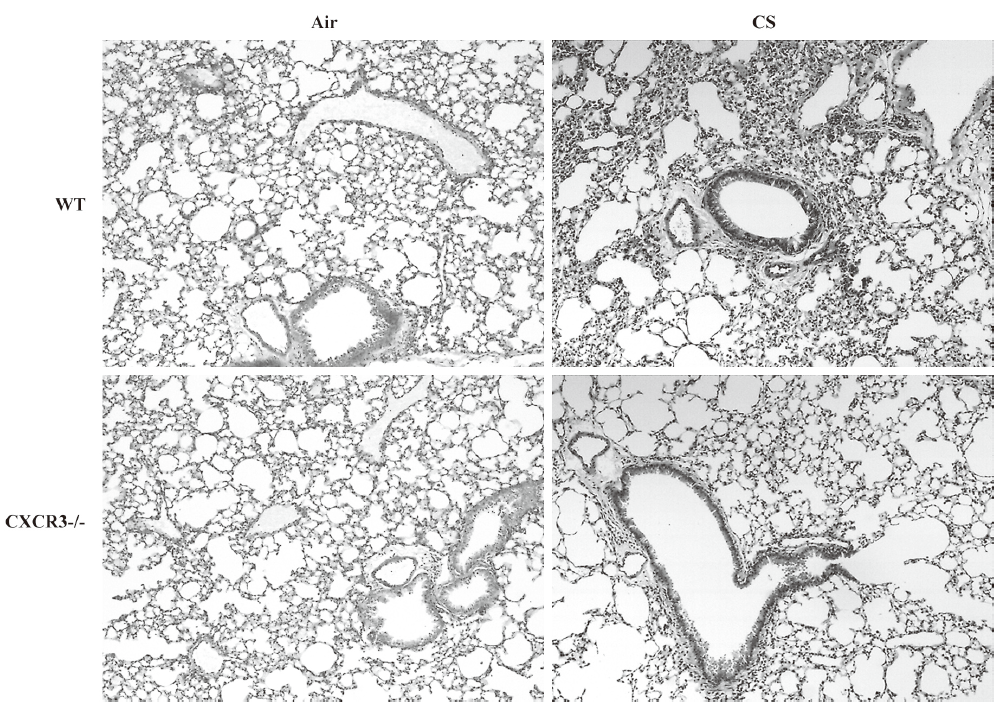
Histological evaluation As shown in Figure 2, there were less leukocytes accumulating in the pulmonary parenchyma and peribronchus in CXCR3–/– mice compared with the WT mice. The air-exposed control mice showed none of these changes.
Analysis of mRNA expression of cytokines and chemokines in lung homogenates The mRNA expression of TNF-α, IFN-γ, KC (the murine homolog for IL-8), and TGF-β was markedly decreased in the lungs from CXCR3–/– mice compared with WT mice at 2 h after the last CS exposure (Figure 3A–3D).
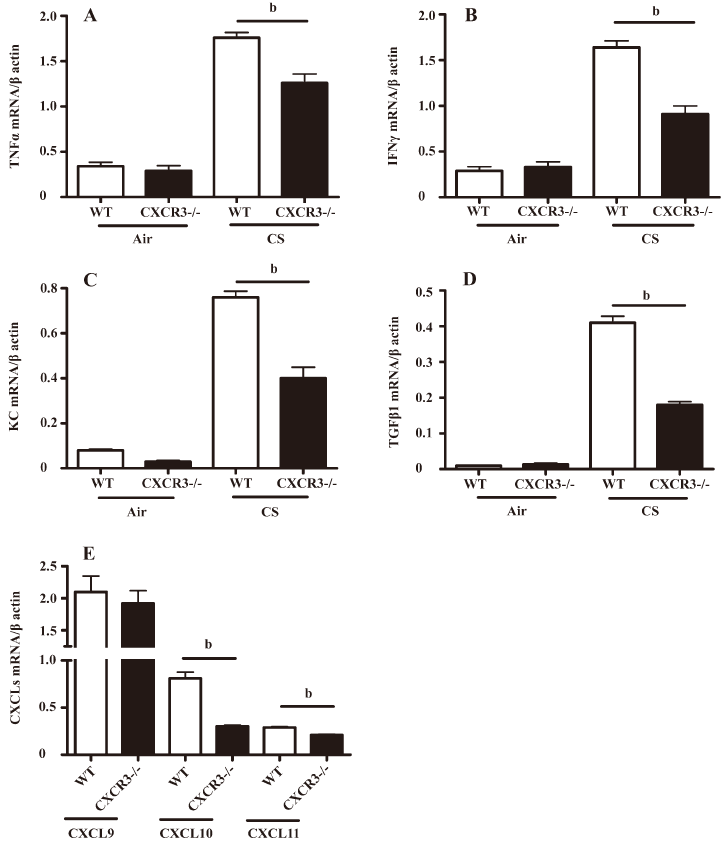
We also observed that the mRNA expression of IFN-γ-inducible CXCL10 and CXCL11, but not CXCL9, was significantly downregulated at 2 h after CS exposure in the lungs from CXCR3–/– mice relative to CXCR3 WT mice (Figure 3E), while the mRNA expression of the CXCR3 ligands from air-exposed mice was not detectable by RT-PCR.
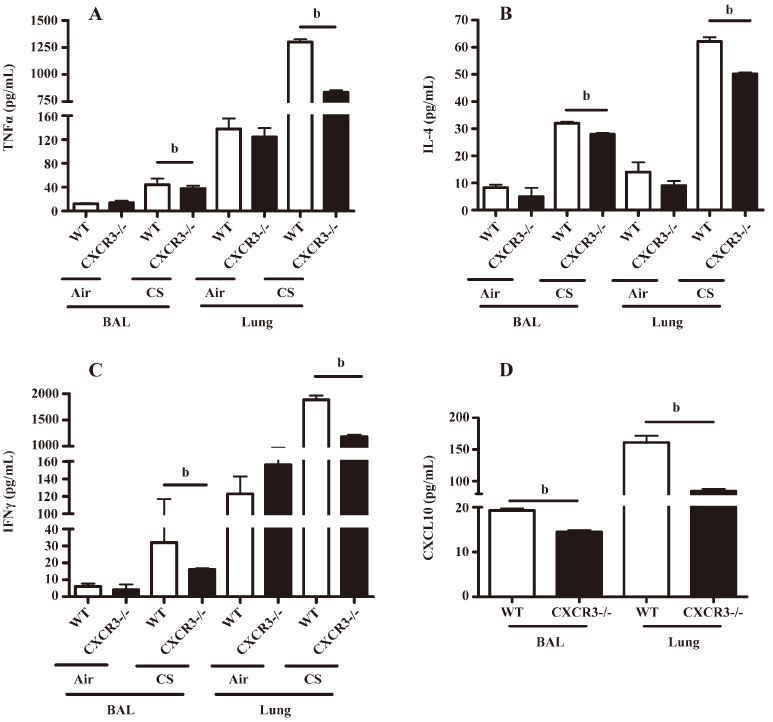
Analysis of inflammatory cytokines and CXCL10 in the BAL fluid and lung homogenates The levels of TNF-α, IL-4, and IFN-γ in the BAL fluid and lung homogenates were significantly lower in the CXCR3–/– mice than in the WT mice (Figure 4A–4C). Importantly, we demonstrated that the CXCL10 concentration in the BAL fluid and lung homogenates was significantly decreased in the CXCR3–/– mice compared with the WT mice at 2 h after CS exposure (Figure 4D). CXCL10 was not detectable in the BAL fluid and lung homogenates in air-exposed mice (data not shown).
Discussion
In this study, we have shown that the deletion of the CXCR3 gene significantly prevented an acute inflammatory response in mice exposed to CS, suggesting that CXCR3 may be a key factor in determining the pathogenesis of the acute lung injury caused by CS through regulating the recruitment of inflammatory cells, particularly CD8+ T cells, into the airways and lungs, initiating the production of inflammatory cytokines and IFN-γ-inducible CXCL10.
The local inflammatory response in COPD is associated with the infiltration of leukocytes, including macrophages, and neutrophils, and T cells into lungs; members of the CXC chemokine family regulate this infiltrating process[10]. Consistent with previous reports of a murine model induced by acute CS exposure[7,9], neutrophils, an invariable component in COPD, represented the majority of cells (approximately 50% of leukocytes in the BAL fluid and lung tissues) at 2 h after 3 consecutive CS exposures in this study. In the CS-exposed CXCR3–/– mice, we observed a significant reduction in the severity of lung inflammation, as evidenced by fewer inflammatory cells, including lymphocytes and neutrophils in the airways and lung tissues. These observations can be explained by the lack of CXCR3, and therefore, points to an important role for CXCR3 in CS-induced lung pathology.
CS has been postulated to induce macrophages, and alveolar epithelial cells secrete chemotactic factors capable of attracting more inflammatory cells into the inflamed sites[11]. We first took the advantage of the gene targeting technique, using CXCR3–/– mice to explore the contribution of CXCR3 to pulmonary inflammation induced by CS. Consistent with a previous study reporting that mice lacking CD8+ T cells were resistant to CS-induced lung inflammation[12], we were able to demonstrate that less CD8+ T cells, as well as CD4+ T cells, were accumulated in the airways and lungs from CXCR3–/– mice. Upon CS insult, the leukocytes infiltrating into the airways and lungs contribute to the inflammation and promote pulmonary injury by secreting a variety of cytokines, such as Th1-secreting cytokine IFN-γ, Th2-releasing cytokine IL-4, pro-inflammatory cytokine TNF-α, and neutrophil chemokine IL-8[13]. We showed that the induction of the inflammatory cytokines (TNF-α, IL-4, and KC) was significantly blunted in both the BAL fluid and the lung homogenates of CXCR3–/– mice exposed to CS compared with the WT mice. These data may reflect the downstream effect of the CXCR3 deficiency, where the blunted pulmonary inflammation further caused the reduced release of inflammatory mediators. The elevated levels of IL-4 and TNF-α in the WT mice may prime CD8+ cytotoxic responses, maintain their memory, and ensure their survival[2]. In particular, we found that the induction of IFN-γ, a powerful inducer for CXC chemokines[14], was significantly inhibited in both the BAL fluid and lung homogenates in CXCR3–/– mice. These data may indicate that CXCR3 mediates the process of pulmonary inflammation via the regulation of the cellular immunological reaction and the production of inflammatory cytokines. In this study, we also observed the early induction of TGF-β1, a profibrotic mediator, at the mRNA level in WT mice compared with CXCR3–/– mice. This finding is consistent with a recent report suggesting increased TGF-β1 gene expression in tracheal explants upon a single CS exposure[15]. Taken together, these data imply that the induction of TGF-β1 will cause the production of TGF-β1 in small airways.
The roles of chemokines and chemokine receptors in the pathogenesis of COPD have been extensively addressed[16–19]. Chemokines participate in the recruitment of a variety of leukocytes via their respective receptors[10]. For example, CXCL9/Mig, CXCL10/IP-10, CXCL11/ITAC, and CCL5/eotaxin, ligands for CXCR3 and CCR5 [chemokine (C-C motif) receptor 5], respectively, were shown to be elevated in sputum from COPD patients compared with non-smokers[20]. CXCL8/IL-8 is a potent chemoattractant and its concentration in sputum and BAL fluid correlates with the increased accumulation of polymorphonuclear neutrophils[13]. Accordingly, chemokine receptors have been implicated in the pathogenesis of lung inflammation and emphysema in rodent models of CS exposure. For example, a small molecule inhibitor of CXCR2 reduced acute pulmonary inflammation, including neutrophilic inflammation induced by CS exposure[9]. In CCR5–/– mice, lung tissue inflammation, apoptosis, and emphysema induced by IFN-γ and CS were significantly decreased, including a decrease in macrophage inflammatory protein (MIP)-1α, MIP-1β, and CXCL10/IP-10[16,17]. Using CCR6–/–mice, CCR6, together with its ligand CXCL20/MIP-3α, is involved in CS-induced lung inflammation and emphysema, and that the interaction between CCR6 and CCL20/MIP-3α could also mediate the accumulation of dendritic cells in the lungs in COPD[18,19].
To our knowledge, our paper is the first to specifically focus on the importance of CXCR3 in the pathogenesis of acute lung inflammation induced by CS by using CXCR3–/– mice. In this investigation, we clearly demonstrated that the level of CXCL10/IP-10 was significantly and consistently elevated at both the mRNA and protein levels in WT mice at 2 h after the last CS exposure compared with CXCR3–/– mice. In contrast, the mRNA expression of CXCL10/IP-10 was seen to be slightly induced in CXCR3–/– mice. CXCR3 is highly expressed on those T lymphocytes that have differentiated to express the Th1 (CD4+) or Tc1 (CD8+) phenotype, suggesting that CXCL10 might be involved in the specific recruitment of leukocyte adhesion, as CXCR3 stimulation increases T-lymphocyte attachment to activated vascular endothelia[21].
In summary, we have shown that CXCR3 regulates acute lung inflammation induced by CS via the recruitment of CD8+ T cells into the lungs to trigger the inflammatory response cascade with the overexpression of some inflammatory cytokines and chemokines. We have determined the importance of CXCR3 in the pathogenesis of CS-induced lung inflammation, and this discovery may provide a therapeutic target for treating CS-associated pathologies.
Acknowledgements
We are grateful to Prof Craig GERARD (Harvard Medical School, Boston, USA) who provided the CXCR3 knockout mice, the staff of the Animal Center-PUMC (Beijing, China) for caring for the animals, and particularly, Hui-min ZHAO. We gratefully acknowledge Prof Richard E RUFFIN and Prof Surendral K BANSAL for critically reading this manuscript and for their helpful suggestions. We also would like to express our thanks to Prof Kian Fan CHUNG for his helpful comments and excellent editing of the manuscript.
References
- Pauwels RA, Rabe KF. Burden and clinical features of chronic obstructive pulmonary disease (COPD). Lancet 2004;364:613-20.
- Chung KF. The role of airway smooth muscle in the pathogenesis of airway wall remodeling in chronic obstructive pulmonary disease. Proc Am Thorac Soc 2005;2:347-54.
- Saetta M, Di Stefano A, Turato G, Facchini FM, Corbino L, Mapp CE, et al. CD8+ T-lymphocytes in peripheral airways of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1998;157:822-6.
- Saetta M, Mariani M, Panina-Bordignon P, Turato G, Buonsanti C, Baraldo S, et al. Increased expression of the chemokine receptor CXCR3 and its ligand CXCL10 in peripheral airways of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2002;165:1404-9.
- Grumelli S, Corry DB, Song LZ, Song L, Green L, Huh J, et al. An immune basis for lung parenchymal destruction in chronic obstructive pulmonary disease and emphysema. PloS Med 2004;1:e8.
- Hancock WW, Lu B, Gao W, Csizmadia V, Faia K, King JA, et al. Requirement of the chemokine receptor CXCR3 for acute allograft rejection. J Exp Med 2000;192:1515-20.
- D’hulst AI, Vermaelen KY, Brusselle GG, Joos GF, Pauwels RA. Time course of cigarette smoke-induced pulmonary inflammation in mice. Eur Respir J 2005;26:204-13.
- Kubo S, Kobayashi M, Masunaga Y, Ishii H, Hirano Y, Takahashi K, et al. Cytokine and chemokine expression in cigarette smoke-induced lung injury in guinea pigs. Eur Respir J 2005;26:993-1001.
- Thatcher TH, McHugh NA, Egan RW, Chapman RW, Hey JA, Turner CK, et al. Role CXCR2 in cigarette smoke-induced lung inflammation. Am J Physiol Lung Cell Mol Physiol 2005;289:L322-8.
- D’Ambrosio D, Mariani M, Panina-Bordignon P, Sinigaglia F. Chemokines and their receptors guiding T lymphocyte recruitment in lung inflammation. Am J Respir Crit Care Med 2001;164:1266-75.
- Barczyk A, Pierzchala W, Kon OM, Cosio B, Adcock IM, Barnes PJ. Cytokine production by bronchoalveolar lavage T lymphocytes in chronic obstructive pulmonary disease. J Allergy Clin Immunol 2006;117:1484-92.
- Maeno T, Houghton AM, Quintero PA, Grumelli S, Owen CA, Shapiro SD. CD8+ T cells are required for inflammation and destruction in cigarette smoke-induced emphysema in mice. J Immunol 2007;178:8090-6.
- Richman-Eisenstat JB, Jorens PG, Hébert CA, Ueki I, Nadel JA. Interleukin-8: an important chemoattractant in sputum of patients with chronic inflammatory airway diseases. Am J Physiol 1993;264:L413-8.
- Wang Z, Zheng T, Zhu Z, Homer RJ, Riese RJ, Chapman HA Jr, et al. Interferon gamma induction of pulmonary emphysema in the adult murine lung. J Exp Med 2000;192:1587-600.
- Churg A, Tai H, Coulthard T, Wang R, Wright JL. Cigarette smoke drives small airway remodeling by induction of growth factors in the airway wall. Am J Respir Crit Care Med 2006;174:1327-34.
- Ma B, Kang MJ, Lee CG, Chapoval S, Liu W, Chen Q, et al. Role of CCR5 in IFN-gamma-induced and cigarette smoke-induced emphysema. J Clin Invest 2005;115:3460-72.
- Bracke KR, D’hulst AI, Maes T, Demedts IK, Moerloose KB, Kuziel WA, et al. Cigarette smoke-induced pulmonary inflammation, but not airway modelling, is attenuated in chemokine receptor 5-deficient mice. Clin Exp Allergy 2007;37:1467-79.
- Bracke KR, D’hulst AI, Maes T, Moerloose KB, Demedts IK, Lebecque S, et al. Cigarette smoke-induced pulmonary inflammation and emphysema are attenuated in CCR6-deficienct mice. J Immunol 2006;177:4350-9.
- Demedts IK, Bracke KR, Van Pottelberge G, Testelmans D, Verleden GM, Vermassen FE, et al. Accumulation of dendritic cells and increased CCL20 levels in the airways of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2007;175:988-1005.
- Costa C, Rufino R, Traves SL, Lapa E, Silva JR, Bames PJ, et al. CXCR3 and CCR5 chemokines in induced sputum from patients with COPD. Chest 2008;133:26-33.
- Barnes PJ, Cosio MG. Characterization of T lymphocytes in chronic obstructive pulmonary disease. PLoS Med 2004;1:e20.