
Hypoxia confers protection against apoptosis via the PI3K/Akt pathway in endothelial progenitor cells
Introduction
Tissue repair and regeneration after injury are thought to involve the selective recruitment of circulating or resident stem cell populations[1]. Endothelial progenitor cells (EPC) are a specific subtype of hematopoietic stem cells that have been isolated from the peripheral blood of humans and express various combinations of antigens traditionally associated with hematopoietic stem cells, as well as endothelial cells [such as CD34, the vascular endothelial growth factor (VEGF) receptor-2, and CD133][2].
Several studies have suggested that EPC play a significant role in the endogenous neovascularization of ischemic tissues and re-endothelization of injured vessels[3]. Moreover, previous studies have demonstrated that hypoxic microenvironments (such as ischemic tissues and injured vessels) are a conditional EPC niche in which many proteins are directly regulated by hypoxia-inducible factor-1 (HIF-1), such as VEGF and the chemokine stromal cell-derived factor-1, which facilitate EPC mobilization, recruitment, and retention at the hypoxic site[4,5]. However, hypoxia on EPC proliferation and survival is not well known.
The phosphatidylinositol 3-kinase (PI3K)/Akt and extracellular signal-regulated protein kinase (ERK) pathways are well-characterized cell survival signaling pathways. Previous studies have demonstrated that ERK and PI3K/Akt are activated by hypoxia in many cell types, such as rat nucleus pulposus cells and human lung cancer cells[6,7], resulting in an anti-apoptotic effect. However, it is not well known whether these pathways are involved in EPC survival.
The goal of the present study was to elucidate the anti-apoptotic roles of the PI3K/Akt and ERK pathways in hypoxia in EPC. Hypoxia suppresses apoptosis induced by serum withdrawal in EPC and activates the PI3K/Akt and ERK pathways. Moreover, blocking PI3K/Akt activation reverses the acquired resistance to serum withdrawal-induced apoptosis in response to hypoxia. To our knowledge, our study is the first to report on the PI3K/Akt pathway in EPC as a survival response to hypoxia.
Materials and methods
Reagents Anti-ERK1/2, anti-HIF-1α, antiphosphorylated ERK1/2, anti-Akt, antiphosphorylated Akt, and the antibodies for the Western blot analysis were purchased from Cell Signaling (Beverly, MA, USA). Antiphosphorylated-glycogen synthase kinase-3β (Ser9) and GSK-3β were purchased from Santa Cruz (Santa Cruz, CA, USA). Annexin V-fluorescein isothiocyanate (FITC) was purchased from BioVision (Mountain View, CA, USA). PI3K inhibitor LY294002 and ERK inhibitor PD98059 were from Cell Signaling (USA). HIF-1α inhibitor YC-1 and mammalian target of rapamycin (mTOR) inhibitor rapamycin were from Alexis Biochemicals (San Diego, CA, USA). Medium 199 was from Sigma (St Louis, MO, USA), EGM-2 MV was from Cambrex Bio Science (Walkersville, MD, USA), and the terminal deoxynucleotidyl transferase-mediated digoxigenin-dUTP nick-end labeling (TUNEL) assay kit was purchased from Roche (China).
Isolation of mononuclear cells and cell culture The EPC were cultured according to previously described techniques[8]. Briefly, mononuclear cells (MNC) were isolated by density centrifugation (Histopaque 1077; Sigma). After purification with 3 washing steps, 105−106 MNC were plated on fibronectin-coated 6-well plates or cell culture flasks. The cells were cultured in endothelial cell basal medium-2 supplemented with EGM-2 MV single aliquots consisting of 20% fetal bovine serum, VEGF, fibroblast growth factor-2, epidermal growth factor, insulin-like growth factor-1, and ascorbic acid. After 4 d in culture, non-adherent cells were removed by washing with phosphate-buffered saline (PBS). New medium was applied, and the culture was maintained through to d 7.
Hypoxic treatment For the hypoxic experiments, cell cultures were placed in a modular incubator chamber (Billups-Rothenberg, Del Mar, CA, USA) and flushed with a gas mixture of 2% O2/5% CO2/93% N2. The airtight chamber containing the cell cultures was incubated for periods of up to 24 h at 37 °C.
Viability assay The cells were plated in 96-well plates (1×105 cells/well) in serum-free medium for 24 h and cultured under hypoxic conditions (2% O2) for 24 h. Cell survival (viability) was measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Briefly, after treatment, MTT diluted in PBS was added to the culture medium to a final concentration of 0.5 g/L. At the end of the incubation period (4 h at 37 °C), the medium was removed and the precipitated formazan crystals were solubilized in DMSO. Product formation was measured by reading the absorbance at 570 nm using a microplate reader (Spectrums Flour Plus; Tecan, Switzerland).
TUNEL TUNEL staining was done according to the manufacturer’s instructions (Roche, China). Briefly, the EPC were incubated in serum-free medium 199 for 24 h and subjected to various treatments for 24 h. The cells were fixed with 4% paraformaldehyde at room temperature. Peroxidase activity in the cells was blocked with 3% H2O2. After being permeabilized with 0.1% Triton X-100, the cells were incubated in 50 μL TUNEL reaction mixture (Roche, China) for 1 h in a humidified atmosphere in the dark, washed 3 times with PBS for 5 min, incubated in 50 μL converter peroxidase (POD) in a humidified chamber at 37 °C for 30 min, washed 3 times with PBS, and then incubated with a diaminobenzidine POD substrate. The cells were washed 3 times and analyzed on a light microscope.
Flow cytometry Fluorescein-conjugated annexin V (Annexin V-FITC) was used to quantitatively determine the percentage of cells undergoing apoptosis. Treated cells were washed twice with cold PBS and then resuspended in binding buffer at a concentration of 5×105 cells/mL. Annexin V-FITC and propidium iodide (PI) were added to the suspended cells. After incubation for 5 min at room temperature in the dark, the percentage of apoptotic cells was analyzed by flow cytometry.
Western blot analysis The EPC were cultured in culture flasks and arrested in serum-free medium 199 for 24 h. After stimulation with hypoxia (2% oxygen) for various periods of time, the cells were washed twice with ice-cold PBS, scraped, and collected. Cell proteins were subjected to SDS-PAGE on 10% polyacrylamide gels and transferred to nitrocellulose membranes. The membranes were incubated overnight with a primary antibody at the appropriate dilution before incubation for 1 h with a secondary antirabbit antibody conjugated to horseradish peroxidase (1:5000). The protein was then visualized using enhanced chemiluminescence solution from Amersham (New Jersey, USA) and X-ray film.
Statistical analysis Data were expressed as mean± SEM for 3–9 individual experiments. The statistical analysis between two groups was performed using unpaired Student’s t-test, and comparisons between multiple groups were made by one-way ANOVA. Probability values were considered significant at P<0.05.
Results
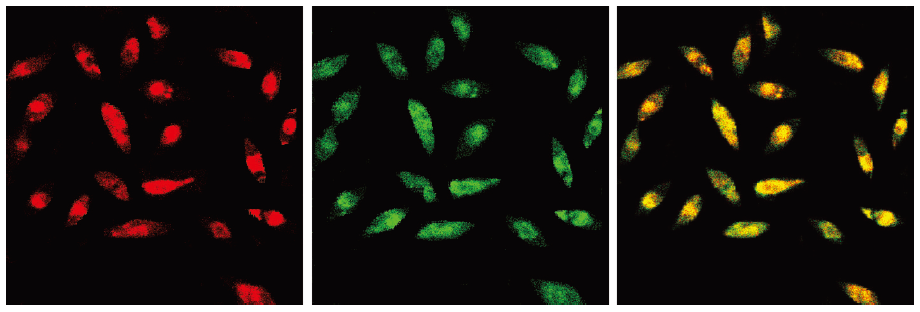
Characterization of EPC After 7 d of culture, expanded ex vivo EPC derived from peripheral blood of healthy human volunteers exhibited spindle-shaped morphology. The EPC were characterized as adherent cells double positive for Dil-Ac-LDL uptake and lectin binding by using laser scanning confocal microscope (LSCM) (Figure 1). A total of 93.8%±4.5% adherent cells showed an uptake of Dil-Ac-LDL and lectin binding after 7 d of culture. This was confirmed by demonstrating the expression of well-established cell surface markers like VE-cadherin (77.3%±6.1%), KDR (78.7%±9.7%), CD34 (31.3%±4.2%), and AC133 (18.7%±5.0%) by fluorescence-activated cell sorting (data not shown).
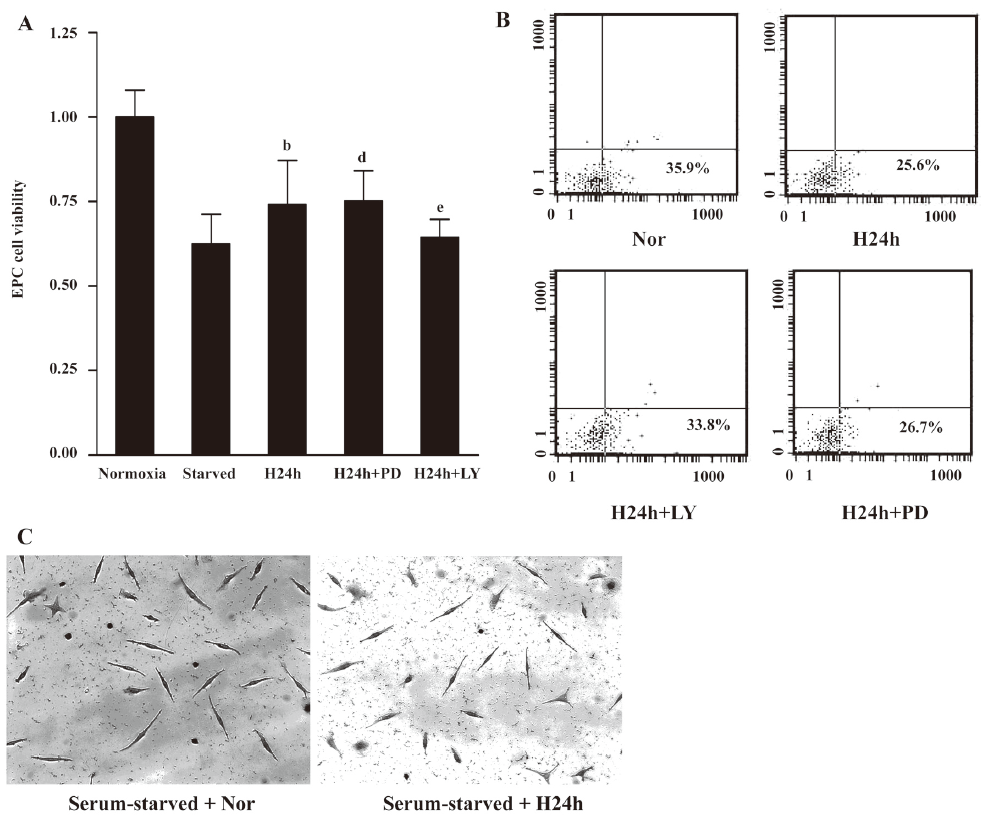
Hypoxia suppressed apoptosis induced by serum withdrawal in EPC To assess the effect of hypoxia on apoptosis induced by serum withdrawal, quiescent EPC were cultured under normoxic or hypoxic conditions (2% O2) for 24 h. Cell viability was measured using MTT assay (Figure 2A). The induction of apoptosis upon serum withdrawal was reduced when cells were cultured under hypoxic conditions. In contrast, if the cells were in normoxia and serum starved, there was a significant decrease in cell survival (P<0.05). The flow cytometric results using Annexin-V-PI supported these observations (Figure 2B). After 24 h in hypoxic environments, 25.6% of cells that were serum starved were in an early apoptotic state. In contrast, under normoxic conditions, serum starvation induced apoptosis of 35.9% for EPC. The enhancement of EPs survival was confirmed by TUNEL assay. The TUNEL assay showed that few nuclei of the hypoxic serum-starved cells were TUNEL positive, whereas serum-starved normoxic cells displayed numerous apoptotic nuclei. These results indicated that hypoxia suppressed serum withdrawal-induced apoptosis in EPC.
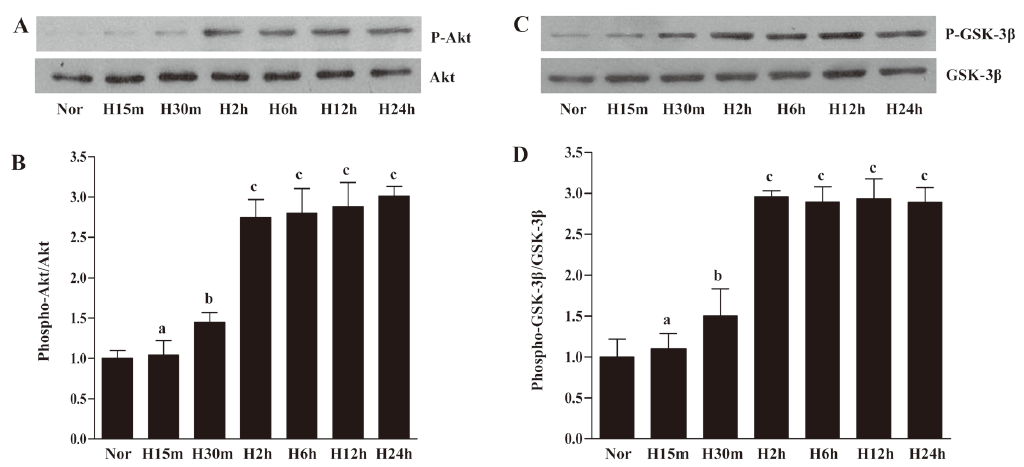
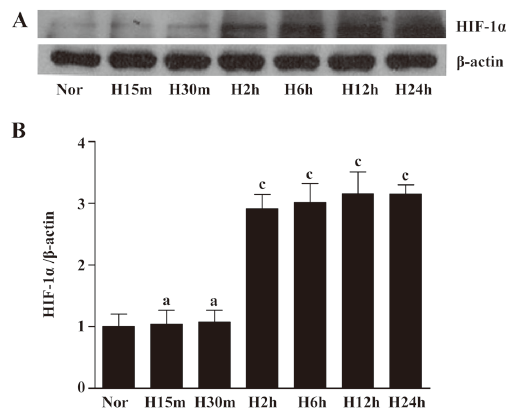
Hypoxia induced Akt and GSK-3β activation in EPC Although many studies have suggested that the activation of Akt is involved in hypoxia-mediated responses, there is little direct evidence for this pathway in EPC under hypoxic conditions. We performed time-course experiments in which quiescent EPC were exposed to hypoxia for different times. We analyzed phosphorylation of Akt-Ser473 and found that Akt phosphorylation at Ser473 significantly increased after 30 min and stayed at a steady level for up to 12 h before reaching maximal levels after 24 h of hypoxic exposure. Total Akt levels remained unchanged under hypoxia (Figure 3A, 3B). Since Akt phosphorylated multiple target proteins, we evaluated the phosphorylation status of GSK-3β, an important effector protein involved in the regulation of apoptosis. We observed that the activation of GSK-3β significantly increased after 30 min under hypoxic conditions and stayed at a steady level for up to 24 h (Figure 3C, 3D). To investigate whether the hypoxic treatment was adequate, HIF-1α expression was determined by Western blotting (Figure 4). HIF-1α significantly increased after 2 h under hypoxic conditions and stayed at a steady level for up to 24 h in EPC. These observations suggested that hypoxia activated these pathways in EPC.
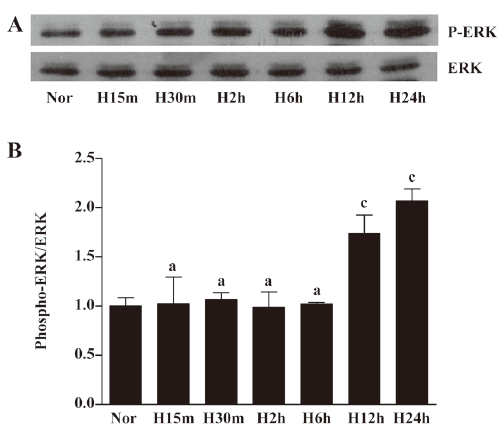
Hypoxia induced ERK activation in EPC To determine whether hypoxia-induced survival was associated with specific change in mitogen-activated protein kinases, the cells were exposed to hypoxia for different times. The activation of this pathway was determined by Western blotting against active ERK (Figure 5). The active phosphorylated form of ERK (Tyr202/Thr204) increased after 12 h of incubation under hypoxia and peaked at 24 h. However, hypoxia did not affect total ERK expression levels.
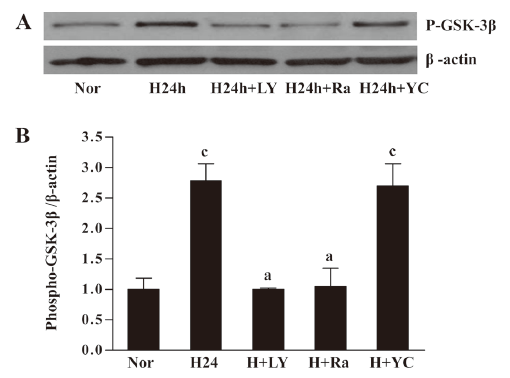
Hypoxia-phosphorylated GSK-3β requires PI3K/Akt activity Intracellular signaling involves a complex network. We were interested in investigating the relationship between the hypoxia-induced activation of Akt and its downstream signaling molecule. We observed that EPC cultured in hypoxia for 24 h showed inactivation (phosphorylation) of GSK-3β. The hypoxia-induced phosphorylation of GSK-3β was abolished by pretreatment with LY294002. The mTOR (the downstream of PI3K/Akt) inhibitor rapamycin (10 μg/L) could block the phosphorylation of GSK-3β. However, the HIF-1α inhibitor YC-1 (50 μmol/L) had no significant effect. These results suggested that GSK-3β might be downstream of PI3K/Akt/mTOR (Figure 6).
Inhibition of apoptosis by hypoxia requires PI3K/Akt activity To evaluate whether the activation of the PI3K/Akt or ERK pathways is necessary for the hypoxia-induced suppression of apoptosis, we investigated the effect of blocking these pathways individually in cells exposed to hypoxia. After incubation for 30 min with or without PD98059 (20 μmol/L) and LY294002 (20 μmol/L), the EPC were cultured in a hypoxic environment for 24 h. The MTT assay and flow cytometric analysis showed that the protective effect under hypoxia could be suppressed by LY294002 (Figure 2A, 2B). These results indicated that protection against cell apoptosis by hypoxia was associated with the activation of the PI3K/Akt survival pathway.
Discussion
In this study, we have shown that hypoxia suppresses serum withdrawal-induced apoptosis in EPC. This effect was paralleled by the promotion of Akt and GSK-3β phosphorylation. Thus, it was likely that the activation of the PI3K/Akt/GSK-3β pathway by hypoxia might be the mechanism responsible for its anti-apoptotic effect.
Recent studies have shown that many intracellular signaling pathways are activated in response to hypoxia, such as protein kinase C, NF-κB, mitogen-activated protein kinase/ERK, and PI3K/Akt signaling pathways. Among these signals, Akt and ERK activation deserve special attention as anti-apoptotic signals. The present study showed that hypoxia activated PI3K/Akt, which concurs with previous reports on PC12 cells, human microvascular endothelial cells, and rat aortic endothelial cells[9–11]. However, our results differed from another investigation that found that hypoxia inhibited PI3K/Akt activation in vascular smooth cell lines[12]. Thus, the effect of hypoxia on PI3K/Akt activation may be cell type specific. We also found a significant increase in the levels of phosphorylated ERK after 12 h under hypoxic conditions. High expression was maintained throughout the culture period. To evaluate whether the activation of the PI3K/Akt and ERK pathways is involved in the hypoxia-induced suppression of apoptosis, we examined the effect of blocking these pathways. LY294002 and PD98059 were used to block the PI3K/Akt and ERK pathways. Interestingly, LY294002 pretreatment inhibited protection against cell apoptosis by hypoxia. ERK inhibitor PD98059 had no significant effect on hypoxia-induced cell survival. These results indicated that protection against cell apoptosis by hypoxia was associated with the activation of the PI3K/Akt survival pathway.
Previous studies have shown that many downstream targets of Akt are involved in cell survival pathways, such as GSK-3, eNOS, BAD, NF-κB, and the FKHR family of forkhead transcriptional factors[13–17]. GSK-3β is a serine/threonine protein kinase. Unlike most other protein kinases, GSK-3β is inactive when phosphorylated. GSK-3β controls several downstream transcription factors that are crucial in cell survival and function, including HIF-1, β-catenin, cyclic adenosine monophosphate (cAMP)-response element-binding protein, AP-1 and cyclin D1[18–21]. EPC transduced with the inactive GSK-3β gene could increase the secretion of angiogenic cytokines (VEGF and interleukin-8) and enhance survival, proliferation, and differentiation[22]. Several upstream kinases have been identified that are capable of phosphorylating GSK-3β, including members of the protein kinase C family and protein kinase A, but Akt is the primary kinase responsible for the phosphorylation and inactivation of GSK-3β[23,24]. In the present study, we showed that GSK-3β could be inactivated by hypoxia in EPC, and the hypoxia-induced phosphorylation of GSK-3β could be abolished by pretreatment with LY294002. These findings suggested that GSK-3β might be downstream of PI3K/Akt in EPC. Interestingly, we found that mTOR inhibitor rapamycin could inactivate GSK-3β. Thus, mTOR might be upstream of GSK-3β. HIF-1α has been regarded as the key mediator of cellular responses to reduced oxygen availability[25]. Some studies have shown that the activation of PI3K/Akt may lead to the inhibition of GSK-3 activity, which then cannot phosphorylate HIF-1α and target it for proteasomal degradation[26,27]. We found that HIF-1α inhibitor YC-1 had no significant effect on the hypoxia-induced phosphorylation of GSK-3β, which could be explained by the fact that HIF-1α may be downstream of GSK-3β in EPC. Further studies are required to elucidate our speculation.
In summary, the present investigation demonstrates that hypoxia suppresses serum withdrawal-induced apoptosis in EPC. Apoptosis suppression by hypoxia correlates with the promotion of Akt and GSK-3β phosphorylation. These results may provide a mechanism by which EPC adapt to their ischemic environments.
References
- Kollet O, Shivtiel S, Chen YQ, Suriawinata J, Thung SN, Dabeva MD, et al. HGF, SDF-1, and MMP-9 are involved in stress-induced human CD34+ stem cell recruitment to the liver. J Clin Invest 2003;112:160-9.
- Urbich C, Dimmeler S. Endothelial progenitor cells: characterization and role in vascular biology. Circ Res 2004;95:343-53.
- Dzau VJ, Gnecchi M, Pachori AS, Morello F, Melo LG. Therapeutic potential of endothelial progenitor cells in cardiovascular diseases. Hypertension 2005;46:7-18.
- Asahara T, Takahashi T, Masuda H, Kalka C, Chen D, Iwaguro H, et al. VEGF contributes to postnatal neovascularization by mobilizing bone marrow-derived endothelial progenitor cells. EMBO J 1999;18:3964-72.
- Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME, et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med 2004;10:858-64.
- Risbud MV, Fertala J, Vresilovic EJ, Albert TJ, Shapiro IM. Nucleus pulposus cells upregulate PI3K/Akt and MEK/ERK signaling pathways under hypoxic conditions and resist apoptosis induced by serum withdrawal. Spine 2005;30:882-9.
- Lee SM, Lee CT, Kim YW, Han SK, Shim YS, Yoo CG. Hypoxia confers protection against apoptosis via PI3K/Akt and ERK pathways in lung cancer cells. Cancer Lett 2006;242:231-8.
- Yamaguchi J, Kusano KF, Masuo O, Kawamoto A, Silver M, Murasawa S, et al. Stromal cell-derived factor-1 effects on ex vivo expanded endothelial progenitor cell recruitment for ischemic neovascularization. Circulation 2003;107:1322-8.
- Alvarez-Tejado M, Naranjo-Suarez S, Jiménez C, Carrera AC, Landázuri MO, del Peso L. Hypoxia induces the activation of the phosphatidylinositol 3-kinase/Akt cell survival pathway in PC12 cells: protective role in apoptosis. J Biol Chem 2001; 276: 22 368–74.
- Robinson MJ, Cobb MH. Mitogen-activated protein kinase pathways. Curr Opin Cell Biol 1997;9:180-6.
- Davis RJ. The mitogen-activated protein kinase signal transduction pathway. J Biol Chem 1993; 268: 14 553–6.
- Loberg RD, Vesely E, Brosius FC III. Enhanced glycogen synthase kinase-3beta activity mediates hypoxia-induced apoptosis of vascular smooth muscle cells and is prevented by glucose transport and metabolism. J Biol Chem 2002; 277: 41 667–73.
- Shiojima I, Walsh K. Role of Akt signaling in vascular homeostasis and angiogenesis. Circ Res 2002;90:1243-50.
- Leinninger GM, Backus C, Uhler MD, Lentz SI, Feldman EL. Phosphatidylinositol 3-kinase and Akt effectors mediate insulin-like growth factor-I neuroprotection in dorsal root ganglia neurons. FASEB J 2004;18:1544-6.
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997;91:231-41.
- Romashkova JA, Makarov SS. NF-kB is a target of AKT in antiapoptotic PDGF signalling. Nature 1999;401:86-90.
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999;96:857-68.
- Wang X, Xiao Y, Mou Y, Zhao Y, Blankesteijn WM, Hall JL. A role for the beta-catenin/T-cell factor signaling cascade in vascular remodeling. Circ Res 2002;90:340-7.
- Fiol CJ, Williams JS, Chou CH, Wang QM, Roach PJ, Andrisani OM. A secondary phosphorylation of CREB341 at Ser129 is required for the cAMP-mediated control of gene expression. A role for glycogen synthase kinase-3 in the control of gene expression. J Biol Chem 1994; 269: 32 187–93
- Boyle WJ, Smeal T, Defize LH, Angel P, Woodgett JR, Karin M, et al. Activation of protein kinase C decreases phosphorylation of c-Jun at sites that negatively regulate its DNA-binding activity. Cell 1991;64:573-84.
- Alt JR, Cleveland JL, Hannink M, Diehl JA. Phosphorylation-dependent regulation of cyclin D1 nuclear export and cyclin D1-dependent cellular transformation. Genes Dev 2000;14:3102-14.
- Choi JH, Hur J, Yoon CH, Kim JH, Lee CS, Youn SW, . Augmentation of therapeutic angiogenesis using genetically modified human endothelial progenitor cells with altered glycogen synthase kinase-3beta activity. J Biol Chem 2004; 279: 49 430–8.
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995;378:785-9.
- Goode N, Hughes K, Woodgett JR, Parker PJ. Differential regulation of glycogen synthase kinase-3 beta by protein kinase C isotypes. J Biol Chem 1992;267:16878-82.
- Sodhi A, Montaner S, Miyazaki H, Gutkind JS. MAPK and Akt act cooperatively but independently on hypoxia inducible factor-1alpha in rasV12 upregulation of VEGF. Biochem Biophys Res Commun 2001;287:292-300.
- Flügel D, Görlach A, Michiels C, Kietzmann T. Glycogen synthase kinase 3 phosphorylates hypoxia-inducible factor 1alpha and mediates its destabilization in a VHL-independent manner. Mol Cell Biol 2007;27:3253-65.
- Mottet D, Dumont V, Deccache Y, Demazy C, Ninane N, Raes M, . Regulation of hypoxia-inducible factor-1alpha protein level during hypoxic conditions by the phosphatidylinositol 3-kinase/Akt/glycogen synthase kinase 3beta pathway in HepG2 cells. J Biol Chem 2003; 278: 31 277–85.