
Effects of breast cancer resistance protein inhibitors and pharmaceutical excipients on decreasing gastrointestinal toxicity of camptothecin analogs1
Introduction
Camptothecin (CPT) is a promising antitumor agent that inhibits nuclear enzyme topoisomerase І[1]. Inhibition of topoisomerase І blocks DNA replication and thus selectively affects cancer cells. The major dose-limiting toxicity of CPT is myelosuppression and delayed-type diarrhea[2,3]. Severe delayed-type diarrhea, however, exhibits a large degree of interpatient variability[4,5] and does not always respond adequately to conventional antidiarrheal agents[6]. For example, irinotecan (CPT-11)-induced delayed-type diarrhea has been reported to be severe (NCI-CTC grade 3–4) in ~25% of patients[7], and less severe diarrhea might even influence continuation of CPT-11 therapy. 10-Hydroxycamptothecin (HCPT), which is commonly used in China, also induces severe diarrhea, and 38% of patients treated with HCPT were associated with delayed-type diarrhea[3]. To date, there is no generally accepted prophylactic treatment for delayed-type diarrhea caused by CPT.
Biliary excretion is a major elimination pathway for CPT and their metabolites. After administration, the parent compound of CPT-11 is mainly excreted in bile both in rats (>32%) and humans (>26%)[8,9]. Its active metabolite SN-38 is further conjugated to SN38-Glu in the liver and mainly excreted via the bile duct[9]. SN38-Glu can be deconjugated by intestinal microflora to regenerate SN-38, which may cause diarrhea[10,11]. As reported[12], 61% of the prototype HCPT was excreted via the bile duct and eliminated from feces within 48 h of administration. The gastrointestinal toxicity of this agent was related to injection dose and the amount of drug excreted[13]. Therefore, it was proposed that the mechanism of toxicity was related to the biliary excretion of CPT and its metabolites[10]. In light of this mechanism, it is likely that inhibition of the biliary excretion of CPT and their metabolites could reduce gastrointestinal toxicity and severe diarrhea. Horikawa[14] reported that coadministration of probenecid reduced the biliary excretion of CPT-11 and its metabolites, and ameliorated the intestinal toxicity provoked by irinotecan. However, long-term coadministration of these micromolecular drugs may cause systemic toxicity to other organs, and their mechanisms of action have not been identified.
The breast cancer resistance protein (BCRP, gene symbol ABCG2) is an ATP-binding cassette (ABC) transporter[15]. It confers high levels of resistance to a wide variety of antitumor drugs including mitoxantrone and the CPT (e.g. topotecan and irinotecan) in several drug resistant cell lines[16,17]. Recent studies have demonstrated that BCRP is expressed in various normal tissues in the body[15]. In particular, BCRP is highly expressed in the apical membrane of small intestine epithelium, in liver canalicular membrane, and in the apical membrane of placental syncytiotrophoblasts. Consistent with this pattern of tissue localization, BCRP has been shown to play a significant role in the absorption, distribution and elimination of BCRP substrates. Therefore, BCRP inhibitors may increase the systemic exposure of drugs that are BCRP substrates[18–20]. Indeed, inhibition of BCRP by GF120918 increased the oral bioavailability of topotecan from 40% to 97% in a clinical study[18]. These data suggest that the highly expressed BCRP in liver canalicular membrane is likely to be the main reason for excessive CPT biliary excretion and their following severe gastrointestinal toxicity. GF120918[21], cyclosporin A[22], and some pharmaceutical excipients[23] (e.g. Pluronic P85 and Tween 20) have been reported to be effective inhibitors of BCRP.
In this study, GF120918, cyclosporin A and some safe pharmaceutical excipients were used to investigate whether these BCRP inhibitors could reduce the biliary excretion of CPT and decrease delayed-type diarrhea rate. Pluronic F68 and PEG stearate are acceptable for pharmaceutical use as solubilizing agents and emulsifying agents. These agents have minor systemic toxicity compared with micromolecular BCRP inhibitors. Whether pharmaceutical excipients can act as BCRP inhibitors and decrease gastrointestinal toxicity is also investigated in this study.
Materials and methods
Materials Hydroxycamptothecin (HCPT) was purchased from Junjie Biotechnology Co, Ltd (Shanghai, China). Irinotecan (CPT-11) was obtained from ZhongShuo Pharmaceutical Technology Development Co, Ltd (Beijing, China). GF120918 was provided by GlaxoSmithKline (Uxbridge, Middlesex, UK). Cyclosporin A (CsA) was obtained from Ruibang Laboratories (Zhejiang, China). Pluronic F68 was a gift from BASF (Germany) and PEG 2000 stearate was purchased from Sigma-Aldrich (St Louis, MO, USA).
Animals Male Sprague–Dawley rats, weighing 200±20 g, were obtained from Shanghai Institute of Materia Medica (Shanghai, China). The room in which the animals were housed was maintained at a temperature of 23±2 °C and a relative humidity of 55%±15%, with a 12-h light-dark cycle. Food and water were freely available throughout the acclimatization and experimental periods. All experiments were carried out under standard guidelines for animal care.
Study of drug biliary excretion Male Sprague–Dawley rats were anesthetized by a single intraperitoneal injection of 20% ethylurethane solution (approximately 0.6 mL/100 g of body weight) without prior fasting. After each rat was anesthetized completely, the abdomen was opened along the longitudinal midline. The liver was checked for any obvious tumors or signs of inflammation. The common bile duct was visualized and separated. A small incision was made on the anterior wall using microscissors. The common bile duct was then cannulated with a fine cannula, 0.50 mm in diameter, and secured into position with a cotton thread. Bile samples were collected in tubes. During the experiments, the rats were placed in restrictive cages under anesthesia and their body temperature was maintained at 37 °C.
The preparations listed in Table 1 were injected intravenously into the tail vein 20 min after bile duct cannulation. Briefly, CPT-11 and HCPT injection were given 10 min after injection of BCRP inhibitor (CsA, GF120918, Pluronic F68 and PEG2000 stearate) solutions. Two types of solutions in each group were injected intravenously from different tail veins of rats. Bile samples were collected every 30 min for 5 h, and their volumes were measured.
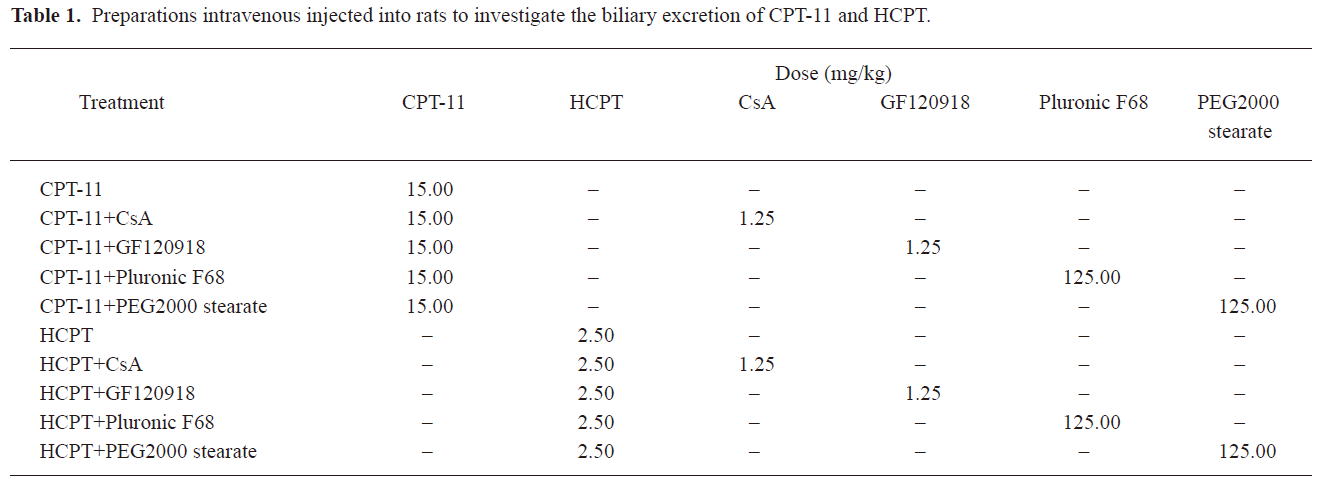
Full table
HPLC analysis of HCPT and CPT-11 in bile The concentrations of HCPT and CPT-11 in bile samples were assayed using reversed-phase HPLC with a LC-20A system (Shimadzu, Japan).
Determination of HCPT concentration was carried out by HPLC as follows: Briefly, the column used was a ZORBAX SB-C18 5 µm, 150×4.6 mm (Agilent, Santa Clara, CA, USA), and the mobile phase consisted of 55:45 mixtures of 50 mmol/L triethylamine-acetate buffer (pH 6.0) and methanol. The flow rate was set at 1.0 mL/min and HCPT was monitored at 382 nm. 200 µL of bile was sampled at selected time intervals and transferred to polypropylene tubes. 10 µL of orthophosphoric acid and 50 µL of CPT (100 µg/mL) as an internal standard were added and stored away from light for 2 h. 1.5 mL of ethyl acetate was added to the samples and extracted for 5 min. The mixture was centrifuged at 5000 r/min for 10 min. The clear supernatant of 1.2 mL was dried under nitrogen and dissolved in 150 µL mobile phase. The solution was centrifuged at 8000 r/min for 15 min prior to HPLC analysis.
The concentration of CPT-11 was determined as follows: The column was a ZORBAX SB-C18 5 µm, 150×4.6 mm (Agilent), and the mobile phase consisted of acetonitrile-50 mmol/L dipotassium hydrogen phosphate buffer containing 10 mmol/L sodium 1-octane-sulfonate, with the pH adjusted to 3.0 with orthophosphoric acid (25/75, v/v). The mobile phase was delivered at a flow-rate of 1.0 mL/min, and the column effluent was monitored at 370 nm. 10 µL of orthophosphoric acid and 50 µL of CPT (100 µg/mL) as an internal standard were added to 200 µL of bile sample and stored away from light at 37 °C for 2 h. 200 µL of ice-cold methanol was added, vortex mixed (5 min) and centrifuged (8000 r/min for 15 min). The clear supernatant (50 µL) was injected onto the HPLC.
Monitoring of CPT-induced diarrhea Sprague-Dawley rats were given CPT-11 (50 mg/kg) and HCPT (10 mg/kg) intravenously into the tail vein once a day for 10 consecutive days according to the published reports[24]. In the BCRP inhibitor group, 1.25 mg/kg of GF120918 and 125 mg/kg of PEG 2000 stearate were coadministered, respectively. Diarrhea observed after the final administration (beginning from d 5) was considered delayed-type diarrhea. The severity of diarrhea was scored according to the following standard as described previously:
• Normal (0, normal stool)
• Slight (1, slight wet stool without staining of the coat)
• Moderate (2, wet and unformed stool with moderate perianal staining of the coat)
• Severe (3, watery stool with severe staining of the coat around the anus)
The mean score and the body weight of rats each day were calculated.
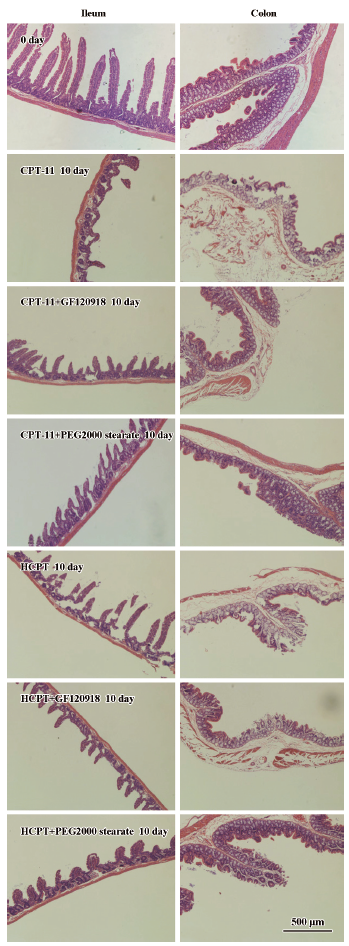
Histological evaluation of intestinal damage The effects of coadministered GF120918 and PEG 2000 stearate on CPT-induced intestinal epithelial injuries in rats were evaluated by examining the histological changes at the microscopic level. The experimental Sprague–Dawley rats were killed on d 10, and the intestinal tissues (ileum, cecum and colon) were collected and fixed in 10% neutral buffered formaldehyde. After routine processing, segments were embedded in paraffin wax using a paraffin embedding machine (Thermo Shandon Ltd). Slices of 4 µm were obtained using a rotary microtome (Leica RM2135, Leica Microsystem AG, Wetzlar, Germany), stained with hematoxylin-eosin (H&E) and subjected to light microscopic examination (Olympus Microscope, Tokyo, Japan). All microscopic and histological assessments were carried out using a blind method to prevent observer bias.
Statistical analysis All data represent at least 3 independent experiments and are expressed as the mean±SD unless otherwise indicated. Statistical comparisons for multiple groups were carried out using a one-way ANOVA followed by comparisons using the Origin 6.0 program. Differences between the 2 groups were analyzed using unpaired Student’s t test. The probability of P<0.05 and P<0.01 were considered to be statistically significant.
Results
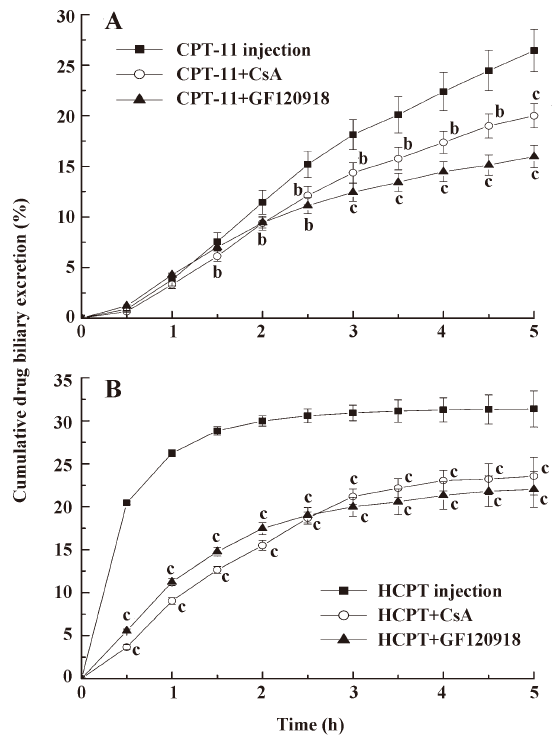
Effects of micromolecular BCRP inhibitors on the biliary excretion of CPT CsA and GF120918 were intravenously injected into the tail vein of rats to investigate whether BCRP affects biliary excretion of CPT. The results (Figure 1) showed that the biliary excretion of CPT-11 and HCPT was significantly reduced following coadministration of CsA and GF120918 (P<0.05, P<0.01). The cumulative biliary excretion of CPT-11 in 5 h was decreased from 26.5% to 20.0% and to 15.9% after coadministration of CsA and GF120918, respectively. HCPT cumulative biliary excretion was decreased from 31.4% to 23.6% and to 22.0%, respectively. The cumulative volumes of biliary excretion have no significant differences among these groups (data not shown). This indicated that CsA and GF120918 decreased the biliary excretion rate and concentration of CPT-11, and inhibited the burst excretion of HCPT within 2 h of administration. The concentrations of CPT-11 and HCPT in intestine were thus diminished following coadministration of CsA and GF120918.
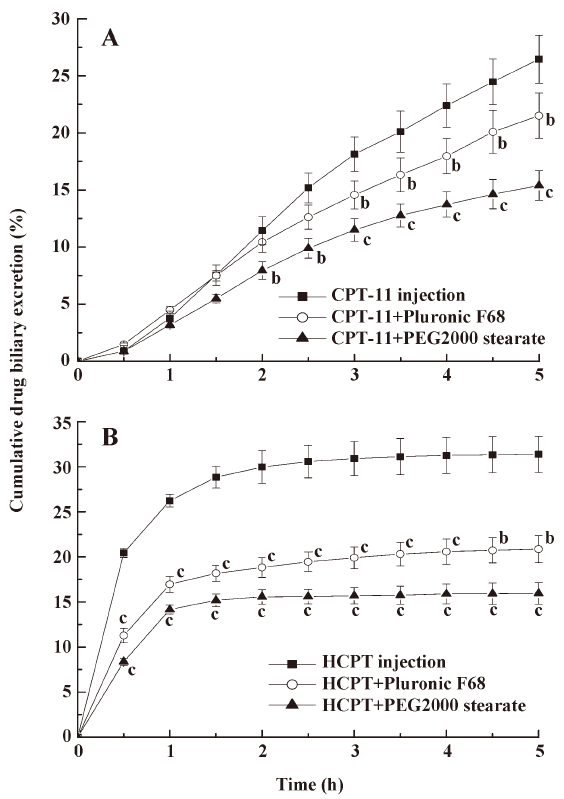
Effects of pharmaceutical excipients on the biliary excretion of CPT Whether pharmaceutical excipients can be used as BCRP inhibitors and reduce the biliary excretion of CPT-11 and HCPT was considered in this study. After coadministration of 150 mg/kg Pluronic F68 and PEG 2000 stearate with CPT-11 and HCPT, bile samples from the rats were collected and determined at designated times (Figure 2). The concentration of CPT (CPT-11 or HCPT) in biliary excretion after coadministration with Pluronic F68 and PEG 2000 stearate were significantly lower than non-excipient-treated groups (P<0.05, P<0.01), and the biliary excretion volumes of these groups were almost the same. The results revealed that the excipients decreased CPT biliary excretion similar to that of the BCRP inhibitors (GF120918 and CsA). PEG 2000 stearate was more effective in decreasing biliary excretion than Pluronic F68.
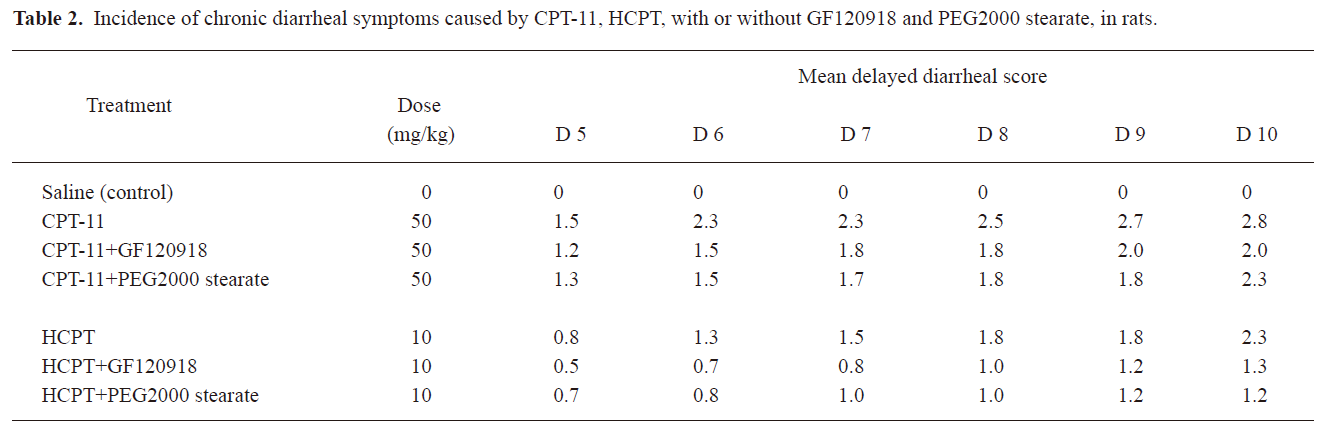
Effects of BCRP inhibitors on CPT-induced delayed diarrhea in rats GF120918 and PEG 2000 stearate have been reported as BCRP inhibitors and were found to reduce the biliary excretion of CPT-11 and HCPT in the above experiments. In addition, these agents were more efficient than CsA and Pluronic F68. Therefore, we examined GF120918 and PEG 2000 stearate as BCRP inhibitors to demonstrate the importance of biliary excretion in delayed-type diarrhea induced by CPT-11 and HCPT in rats. Injection of CPT-11 (20 mg/kg) alone resulted in severe (grade 2–3) delayed diarrhea from d 5 to d 10 (Table 2). When GF120918 and PEG 2000 stearate were coadministered, a diarrheal score of 1–2 was observed (Table 2). The diarrheal score for CPT-11+GF120918 was not obviously different from that for CPT-11+PEG 2000 stearate, but was significantly lower than that for CPT-11 alone. Administration of HCPT caused delayed diarrhea (grade 1–2), which occurred from d 6 to d 10. A lower diarrheal score of around 1 was observed following coadministration of GF120918 and PEG 2000 stearate; however, no significant differences were observed.
Full table
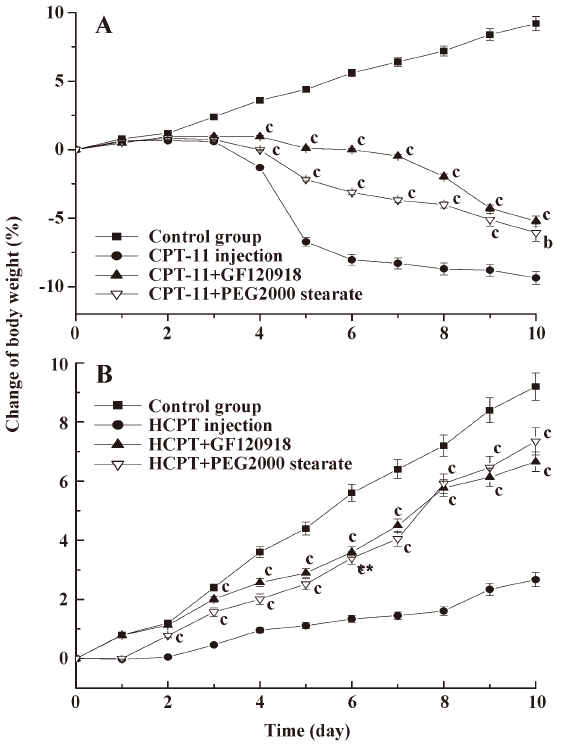
In the control group, the body weight increased continuously, whereas the body weight of rats treated with CPT-11 injection started to fall at d 3–4, and this reduction was more marked at d 5 (Figure 3). Compared with the CPT-11 alone injection group, coadministration of GF120918 and PEG 2000 stearate with CPT-11 significantly ameliorated the body weight loss (P<0.01). Although the body weight of rats in the HCPT injection group increased within 10 d, the growth rate was dramatically lower than that in the groups coadministered with GF120918 and PEG 2000 stearate (P<0.01). There were no significant differences between the HCPT+GF120918 and the HCPT+PEG 2000 stearate groups.
BCRP inhibitors reduced CPT-induced intestinal mucous membrane damage Marked microscopic pathological differences were observed in the intestinal tissues of rats receiving CPT-11 and HCPT injection with or without BCRP inhibitors (Figure 4). Administration of CPT-11 and HCPT resulted in severe damage to the intestinal mucosal membrane which was characterized by stubby villi and patches without villi, goblet cell mucus depletion, destruction of normal mucosal architecture, epithelial vacuolation, degeneration of crypts, crypt abscesses and inflammatory cellular infiltration. However, gastrointestinal toxicity in rats coadministered with GF120918 and PEG 2000 stearate decreased significantly
Discussion
In the present study, we found that coadministration of CsA and GF120918 could reduce the biliary excretion concentration of CPT-11 and HCPT. It is worth mentioning that GF120918 reduced the biliary excretion of CPT-11 and HCPT more than CsA (Figure 1). It is reasonable that GF120918 (IC50=0.3 μmol/L) was more efficient in inhibiting BCRP compared with CsA (IC50=26.1 μmol/L). The actions of these BCRP inhibitors have also been shown to be dose-dependent[22]. GF120918 and CsA were administered at the same dose in our experiments. Thus, GF120918 displayed more efficiency than CsA in inhibiting BCRP. Although GF120918 and CsA are effective inhibitors of both BCRP and P-gp, CPT such as topotecan, CPT-11, and SN-38 are poor P-gp substrates [25,26]. In contrast, CPT are high affinity substrates of BCRP. Therefore, the inhibitory effects of GF120918 and CsA on BCRP are presumed to influence the metabolic processes of CPT-11 and HCPT. Coadministration of BCRP inhibitors will result in reduced biliary excretion of CPT.
The result in Figure 2 shows that Pluronic F68 and PEG 2000 stearate could reduce the biliary excretion of CPT-11 and HCPT. Yamagata[27] has reported on the inhibitory effects of fifteen excipients on BCRP, Pluronic P85 and polyoxyethylene stearate significantly increased the uptake of mitoxantrone in BCRP overexpressed MDCK cells. The coadministration of Pluronic P85 and Tween 20 improved the oral bioavailability of BCRP substrates by inhibiting BCRP function in the small intestine[23]. These studies demonstrated that some excipients such as Pluronic and polyoxyethylene stearate might be potent BCRP inhibitors. The exact mechanism by which excipients inhibit efflux transporters is unknown. It has been reported that Pluronic P85 significantly reduces intracellular ATP levels, and this correlates well with the inhibition of efflux transporters[28]. The results suggest that pharmaceutical excipients can decrease the biliary excretion of CPT-11 and HCPT due to inhibition of BCRP in liver canalicular membrane. In addition, these excipients or surfactants are commonly used in human pharmaceutical formulations and are considered safe and relatively non-toxic. The use of excipients as inhibitors of BCRP may lead to important pharmacotherapeutic benefits.
It has been proposed that the severe gastrointestinal toxicity induced by CPT-11 and HCPT results from exposure of intestinal tissues to SN-38 and HCPT, and this is due to the excessive biliary excretion of CPT. We investigated a potential strategy to inhibit biliary excretion and to reduce exposure of the intestines to CPT. The observed gastrointestinal toxicity, assessed as the diarrheal score and body weight change, was ameliorated by coadministration of GF120918 and PEG 2000 stearate compared with injection of CPT-11 and HCPT alone (Table 2, Figure 3). This was consistent with the results on biliary excretion (Figures 1, 2). CPT-11 induced more severe body weight loss than HCPT. This is probably because the CPT-11 metabolites SN-38 and SN-38G caused more severe cell damage on the intestinal mucous membrane than HCPT. Similar results have also been obtained in clinical studies[12,29,30].
The histological evaluation of intestinal damage revealed that coadministration of GF120918 and PEG 2000 stearate could reduce CPT-induced intestinal mucous membrane damage. The mechanisms for CPT-11- and HCPT-induced intestinal mucous membrane damage are unclear, but appear to be associated with intestinal exposure to these drugs, which induce epithelial apoptosis characterized by the generation of DNA fragments through the action of endogenous endonucleases. These drugs cause severe intestinal inflammatory damage with hypersecretion of pro-inflammatory cytokines. The homeostatic balance between proliferation and apoptosis is essential for intestinal epithelium to function as a physiological and structural barrier. Disruption of this balance leads to villus atrophy and loss of normal absorptive function. Cytotoxic drugs (e.g. CPT-11 and HCPT) can disrupt this mucosal barrier with increased epithelial apoptosis, and cause severe intestinal mucosal membrane damage. Coadministration of GF120918 and PEG 2000 stearate can inhibit BCRP function and decrease the biliary excretion of CPT-11 and HCPT, subsequently diminishing epithelial apoptosis of the intestinal mucosal membrane.
Conclusion
The present study demonstrates that BCRP exhibits important effects on the biliary excretion and gastrointestinal toxicity of CPT. Coadministration of BCRP inhibitors such as GF120918 and CsA reduced the biliary excretion of CPT-11 and HCPT. Pharmaceutical excipients such as Pluronic F68 and PEG 2000 stearate also showed inhibitory effects on BCRP and similarly reduced CPT biliary excretion. The observed gastrointestinal toxicity, assessed as the diarrheal score, body weight change and microscopic pathological damage was ameliorated following coadministration of the BCRP inhibitors and excipients compared with injection of CPT-11 and HCPT alone. The use of excipients as inhibitors of BCRP is safe and relatively non-toxic, and may lead to important pharmacotherapeutic benefits by decreasing the gastrointestinal toxicity of CPT.
References
- Jaxel C, Kohn KW, Wani MC, Wall ME, Pommier Y. Structure-activity study of the actions of camptothecin derivatives on mammalian topoisomerase I: Evidence for a specific receptor site and for a relation toantitumour activity. Cancer Res 1989;49:1465-69.
- Atsumi R, Suzuki W, Hakusui H. Identification of the metabolites of irinotecan, a new derivative of camptothecin, in rat bile and its biliary excretion. Xenobiotica 1991;21:1159-69.
- Yao Y, Sun YJ, Zhao H, Guo YW, Lin F, Cai X, et al. Short-term effects of chemotherapy combined with hydroxycamptothecine and oxaliplatin in treatment of recurrent and metastatic colorectal cancer. Chin J Cancer 2006;8:1035-8.
- Kudoh S, Fukuoka M, Masuda N, Yoshikawa A, Kusunoki Y, Mastui K, et al. Relationship between the pharmacokinetics of irinotecan and diarrhea during combination chemotherapy of cisplatin. Jpn J Cancer Res 1995;86:406-13.
- Negoro S, Fukuoka M, Masuda N, Takada M, Kusunoki Y, Mastui K, et al. Phase I study of weekly intravenous infusion of CPT-11, a new derivative of camptothecin, in the treatment of advanced non-small-cell lung cancer. J Natl Cancer Inst 1991;83:1164-8.
- Takasuna K, Kasai Y, Kitano Y, Mori K, Kakihata K, Hirohashi M, et al. Study on the mechanisms of diarrhea induced by a new anticancer camptothecin derivative, irinotecan hydrochloride (CPT-11), in rats. Folia Pharmacol Jpn 1995;105:447-60.
- Creemers GJ, Lund B, Verweij J. Topoisomerase I inhibitors: topotecan and irinotecan. Cancer Treat Rev 1994;20:73-96.
- Kaneda N, Yokokura T. Nonlinear pharmacokinetics of CPT-11 in rats. Cancer Res 1990;50:1721-5.
- Lokieo F, Canal P, Gay C, Chatelut E, Armand JP, Roche H, et al. Pharmacokinetics of irinotecan and its metabolites in human blood, bile, urine. Cancer Chemother Pharmacol 1995;36:79-82.
- Gupta E, Lestingi TM, Mick R, Ramirez J, Vokes EE, Ratain MJ. Metabolic fate of irinotecan in humans: correlation of glucuronidation with diarrhea. Cancer Res 1994;54:3723-5.
- Takasuna K, Hagiwara T, Hirohashi M, Nomura M, Nagai E, Yokoiu T, et al. Involvement of β-glucuronidase in intestinal microflora in the intestinal toxicity of the antitumor camptothecin derivative irinotecan hydrochloride (CPT-11) in rats. Cancer Res 1996;56:3752-7.
- Zhang L, Li S, Liao H, Jiang WQ, Guan ZZ. Phase I trial of pharmacokinetics and human tolerability to 10-hydroxycamptothecin in patients with advanced malignancy. Chin J Cancer 2001;12:1391-5.
- Huang HQ, Jiang WQ, Hu XH, Lin XB, Liu KF, Li YH, et al. Preliminary study of lyophilized 10-hydroxycamptothecin in advanced or recurrent malignancies. Chin J Cancer 2003;12:1334-8.
- Horikawa M, Kato Y, Sugiyama Y. Reduced gastrointestinal toxicity following inhibition of the biliary excretion of irinotecan and its metabolites by probenecid in rats. Pharm Res 2002;19:1345-53.
- Herwaarden AE, Schinkel AH. The function of breast cancer resistance protein in epithelial barriers, stem cells and milk secretion of drugs and xenotoxins. Trends Pharmcol Sci 2006;27:10-8.
- Doyle LA, Ross DD. Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2). Oncogene 2003;22:7340-58.
- Litman T, Brangi M, Hudson E, Fetsch P, Abati A, Ross DD, et al. The multidrug-resistant phenotype associated with overexpression of the new ABC halftransporter, MXR (ABCG2). J Cell Sci 2000;113:2011-21.
- Kruijtzer CMF, Beijnen JH, Rosing H, Bokkel Huinink WW, Schot M, Jewell RC, et al. Increased oral bioavailability of topotecan in combination with the breast cancer resistance protein and p-glycoprotein inhibitor GF120918. J Clin Oncol 2002;20:2943-50.
- Jonker JW, Smit JW, Brinkhuis RF, Maliepaard M, Beijnen JH, Schellens JH, et al. Role of breast cancer resistance protein in the bioavailability and fetal penetration of topotecan. J Natl Cancer Inst 2000;92:1651-6.
- Jonker JW, Buitelaar M, Wagenaar E, Van Der Valk MA, Scheffer GL, Scheper RJ, et al. The breast cancer resistance protein protects against a major chlorophyll-derived dietary phototoxin and protoporphyria. Proc Natl Acad Sci USA 2002;99:15649-54.
- Hyafil F, Vergely C, Du Vignaud P, Grand-Perret T. In vitro and in vivo reversal of multidrug resistance by GF120918, an acridonecarboxamide derivative. Cancer Res 1993;53:4595-602.
- Gupta A, Dai Y, Vethanayagam RR, Hebert MF, Thummel KE, Unadkat JD, et al. Cyclosporin A, tacrolimus and sirolimus are potent inhibitors of the human breast cancer resistance protein (ABCG2) and reverse resistance to mitoxantrone and topotecan. Cancer Chemother Pharmacol 2006;58:374-83.
- Yamagata T, Kusuhara H, Morishita M, Takayama K, Benameur H, Sugiyama Y. Improvement of the oral drug absorption of topotecan through the inhibition of intestinal xenobiotic efflux transporter, breast cancer resistance protein, by excipients. Drug Metab Dispos 2007;35:1142-8.
- Takasuma K, Hagiwara T, Watanabe K, Onose S, Yoshida S, Kumazawa E, et al. Optimal antidiarrhea treatmeat for antitumor agent irinotecan hydrochloride (CPT-11)-induced delayed diarrhea. Cancer Chemother Pharmacol 2006;58:494-503.
- Hendricks CB, Rowinsky EK, Grochow LB. Effect of P-glycoprotein expression on the accumulation and cytotoxicity of topotecan (SK&F 104864), a new camptothecin analogue. Cancer Res 1992;52:2268-78.
- Brangi M, Litman T, Ciotti M, Nishiyama K, Kohlhagen G, Takimoto C, et al. Camptothecin resistance: role of the ATP-binding cassette (ABC), mitoxantrone-resistance half-transporter (MXR), and potential for glucuronidation in MXR-expressing cells. Cancer Res 1999;59:5938-46.
- Yamagata T, Kusuhara H, Morishita M, Takayama K, Benameur H, Sugiyama Y. Effect of excipients on breast cancer resistance protein substrate uptake activity. J Controlled Release 2007;124:1-5.
- Batrakova EV, Li S, Elmquist WF, Miller DW, Alakhov VY, Kabanov AV. Mechanism of sensitization of MDR cancer cells by pluronic block copolymers: selective energy depletion. Br J Cancer 2001;85:1987-97.
- Shimada Y, Yoshino M, Wakui A, Nakao I, Futatsuki K, Sakata Y, et al. Phase II study of CPT-11, a new camptothecin derivative, in metastatic colorectal cancer. J Clin Oncol 1993;11:909-13.
- Fukuoka M, Niitani H, Suzuki A, Motomiya M, Hasegawa K, Nishiwaki Y, et al. A phase II study of CPT-11, a new derivative of camptothecin, for previously untreated nonsmall-cell lung cancer. J Clin Oncol 1992;10:16-20.