
Nitric oxide inhalation inhibits inducible nitric oxide synthase but not nitrotyrosine formation and cell apoptosis in rat lungs with meconium-induced injury1
Introduction
Meconium aspiration syndrome (MAS) remains a significant cause of morbidity and mortality in term newborn infants with intrauterine hyperoxia, despite improvements in obstetric care. The pathophysiology of lung injury in MAS is very complicated. The complex nature of MAS contributes to its poor response to many therapeutic interventions[1–4]. Clinical and experimental evidence suggest that inhaled nitric oxide (NO) reduces pulmonary hypertension, improves systemic oxygenation and inhibits the transendo-thelial migration of activated neutrophils in a variety of pathological conditions of the lungs, including meconium aspiration[3,5,6].
As a free radical, however, NO inhalation is potentially toxic. NO induces apoptosis in various cell lines[7]. NO inhalation can cause damage to the lung tissue through its reaction with superoxide anions, resulting in the production of peroxynitrite anion (ONOO–). Nitrotyrosine is a marker of ONOO– production, which provides evidence for therapy-induced iNO toxicity[8]. A previous study showed that NO inhalation during the early phase of inflammation did not increase, but rather decreased nitrotyrosine formation[8,9]. However, nitrotyrosine formation in meconium-induced acute lung injury (ALI) with or without inhalation of NO has not been investigated. The anti-inflammatory effect of iNO in meconium-induced ALI is still controversial[3].
The aims of our present study were to explore the mechanism of anti-inflammatory effects and to investigate the possible toxic role of iNO in meconium-induced ALI. A rat model of ALI following meconium aspiration was established. The pulmonary inducible NO synthase (iNOS) protein and interleukin (IL)-1β mRNA expression, nitrotyrosine formation, pulmonary apoptosis, as well as lung injury score, broncho-alveolar lavage (BAL) cell count, myeloperoxidase (MPO) activity and malondialdehyde (MDA) level were measured.
Materials and methods
Meconium preparation First-passed meconium was taken from urine-free diapers of healthy term neonates. The meconium was placed in a sterile jar and frozen for less than 7 d and then lyophilized, pooled and diluted with sterile saline to a final concentration of 20% suspension[10]. This meconium slurry was frozen at -70 oC until use.
Animal management Twenty-four 30–45 d old Sprague-Dawley rats weighing 130 –170 g were studied. A rat model of meconium-induced ALI was established as previously described[10,11]. Briefly, rats were anesthetized with sodium pentobarbital (40 mg/kg, ip). After tracheotomy, an endotracheal tube was placed through the incision, and then stabilized. Prior to meconium instillation, rats were given vecuronium bromide (2 mg/kg, ip). After 15 min, the rats were randomized into 3 groups (n=8 each). Sixteen of the rats were instilled intratracheally with meconium (1 mL/kg) followed by a 3 mL bolus of air to disperse the meconium into the lung to establish the model of ALI. Half of the dose was given with the rat lying on one side and the other half with the rat lying on the other side and then randomized into the Mec/saline group, with intratracheal instillation of 1 mL/kg saline, or the Mec/iNO group, with continuous inhalation of NO at 20 μL/L. The remaining 8 rats were given 1 mL/kg sterile saline intratracheally followed by a 3 mL bolus of air as control. At 60 min after surgery, skin and tracheal incisions were closed with a 4–0 nylon suture. The rats in the Mec/saline group and in the control group were allowed to breath spontaneously in room air.
Nitric oxide exposure Immediately after the skin and tracheal incisions were closed, the rats in the Mec/iNO group were housed for 24 h in a sealed Plexiglas chamber that was flushed continuously with a precise dilution of NO gas (800 μL/L) and medical air (4 mL/kg) to achieve a final NO concentration of 20 μL/L NO concentration in the chamber was continuously monitored by a NO/NO2 monitor confirming that the NO concentration was maintained at 20±1 μL/L and that the NO2 concentration was consistently <2 μL/L.
Sample collection and storage The animals were killed 24 h after treatment. The chest was opened by a midline incision. The lungs were isolated and left lung lavage was carried out. Tissue samples from the right lung were obtained. Apoptosis of lung tissue cells was evaluated using electron microscopy and a terminal deoxynucleodityl transferase-mediated dUTP nick-end labeling (TUNEL) assay was carried out.
Bronchoalveolar lavage total cell count The left lung was lavaged using 31 mL aliquots of cold sterile saline following tracheotomy. Each lung was instilled and cleared 3 times. The recovered amount was always more than 90%. A total BAL cell count was obtained within 4 h.
Pulmonary myeloperoxidase activity and malondial-dehyde assay Myeloperoxidase (MPO) activity was assessed as a measure of pulmonary neutrophil influx and activity. Pulmonary MPO activity and malondialdehyde (MDA) levels were assayed in duplicate on homogenized lung samples, according to the manufacturer’s manual. One unit of MPO activity was expressed as units per gram of wet lung tissue. MDA levels were standardized for tissue protein concentration determined by the Bradford protein assay[12].
Reverse transcription-polymerase chain reaction (PCR) analysis of pulmonary IL-1β mRNA expression Total RNA was isolated with Trizol Reagent (Sangon, Shanghai, China) in accordance with the manufacturer’s instructions. Complementary DNA (cDNA) was prepared by reverse transcription (RT) of 10 µg total RNA using oligo dT18 and 200 u MmuLV reverse transcriptase (Sangon) at 37 oC for 90 min according to the manufacturer’s manual. cDNA was used as a template in the PCR, which was run in Tag-polymerase buffer, supplemented with 1.5 mmol/L MgCl2, 5 µg/mL of each primer, 4 µL of 10 mmol/L dNTPs, and 1 U of Tag-polymerase (TaKaRa, Dalian, China). The total reaction volume was 50 µL. The primers used for IL-1β in RT-PCR were coding, 5'-GAA GCT GTG GCA GCT ACC TAT GTC T-3' and uncoding, 5'-CTC TGC TTG AGA GGT GCT GAT GTA C-3', which yielded a PCR product of 520 bp. The primers for β-actin (200 bp) were coding, 5'-ATG CCA ACA CAG TGC TGT CT-3' and uncoding, 5'-CTG CTT GCT GAT CCA CAT CT-3'. Amplification was carried out for 30 cycles at 94 oC for 1 min, 65 oC for 45 s, 72 oC for 45 s, followed by a 7 min extension at 72 oC. Gene expression was determined by the size of the PCR product in 1.6% agorose gels (in Tris-buffer, pH 8.0), stained with 0.1% μg/mL ethidium bromide and observed under ultraviolet light. The data of the IL-1β mRNA levels was normalized against the housekeeping gene β-actin. Molecular weight markers were run in parallel.
Western blot analysis of pulmonary nitrotyrosine formation and inducible nitric oxide synthase expres-sion Frozen lung tissues were homogenized in protein extraction buffer. The homogenate was left on ice for 30 min before centrifugation at 10 000×g. Protein content was estimated[12], and aliquots of 50 µg homogenate protein were run on 10% (w/v) sodium dodecylsulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) gels. Proteins were subsequently transferred to nitrocellulose membranes and stained with Coomassie blue to verify the equivalent transfer of samples. Membranes were blocked by incubation with 3% (w/v) fat dry milk in PBS for 1 h at room temperature, with shaking. Thereafter, the membranes were incubated overnight at 4 oC with either mouse monoclonal anti-nitrotyrosine (Calbiochem-Novabiochem, San Diego, CA, USA) or rabbit polyclonal anti-iNOS or rabbit polyclonal anti-actin (Santa Cruz, Califonia, USA). The primary antibody was used at dilutions from 1:500 to 1:1000. The blots were washed 4 times (10 min each time) with Tris-buffered saline Tween-20 (TBST), comprising 50 mmol/L Tris-HCl (pH 7.6), 150 mmol/L NaCl and 0.1% Triton X-100, and subsequently incubated for 2 h at room temperature in Tris-buffered saline with horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG antibody (1:5000; Pierce, Rockford, IL, USA). Immunoreactive bands were visualized using the enhanced chemiluminescence system (Pierce) and quantified by densitometric analysis using Scion image software (Kodak, EDAS290 Imager Analysis). Results were calculated as the relative ratio of the specific band compared with actin.
Histological examination Pulmonary tissue samples from the right lower lobe were fixed in 10% buffered formalin, embedded in paraffin, and stained with hematoxylin and eosin for determination of the severity of lung injury. Samples were assessed by a blinded pathologist who was unaware of the group of the rats. A score from 0 to 4 represented the percentage of affected area on the lung section (0=0%, 1=1%–25%, 2=26%–50%, 3=51%–75%, and 4=76%–100%) and was assigned for 3 different characteristics: (i) extension of pulmonary leukocyte infiltration; (ii) amount of intra-alveolar leukocytes; and (iii) amount of exudative debris and edema fluid. The calculated total injury score represents the sum of these scores[3].
Quantification of apoptosis in lung tissues by TUNEL The semiquantification of apoptotic cells in lung tissue was carried out by in situ TUNEL using the Apop Tag Peroxidase In Situ Cell Death Detection Kit (Roche Diagnostic GmbH Mannheim, Germany) according to the manufacturer’s protocol. The number of positive signals in each section was evaluated by 3 independent pathologists using light microscopy. The apoptosis index (AI) was calculated. AI is a measure of the number of positive cells in each 100 cells counted in 5 different blocks in the same section[13].
Electron microscopy Small pieces of lung tissue (1 mm3) from the left lower lobe were obtained for ultrastructural analysis of apoptotic pulmonary cell death by electron microscopy.
Statistical analysis Data are expressed as mean±SD. Statistical significance among sample groups was analyzed with ANOVA. The Mann-Whitney test was used to analyze the data from lung injury score. A level of P<0.05 was considered statistically significant.
Results
The meconium-instilled group showed increased BAL cell count, pulmonary MPO activity and lung injury score when compared with the control group (all P<0.01; Table 1). The expression of IL-β mRNA and iNOS protein was also increased significantly (both P<0.01; Figures 1,2). Pulmonary nitrotyrosine, a marker of ONOO– formation as an index of pulmonary peroxidation, and MDA, a marker of lipid peroxidation, were increased significantly as well (both P<0.01; Table 1; Figure 3). Pulmonary AI, demonstrated by TUNEL staining, was significantly elevated at 24 h after the treatment (P<0.01; Figure 4). This was confirmed by the electron microscopic finding that apoptotic change occurred in pulmonary epithelium in the meconium-instilled rat lungs (Figure 5).
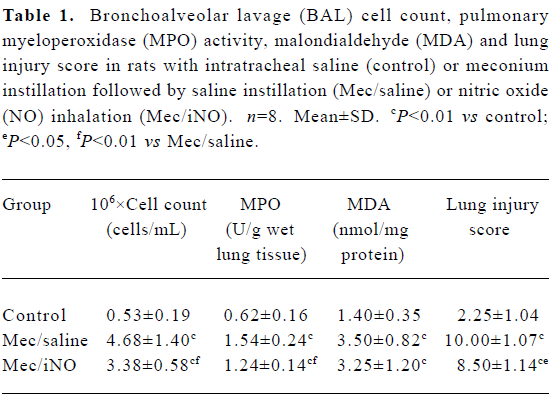
Full table
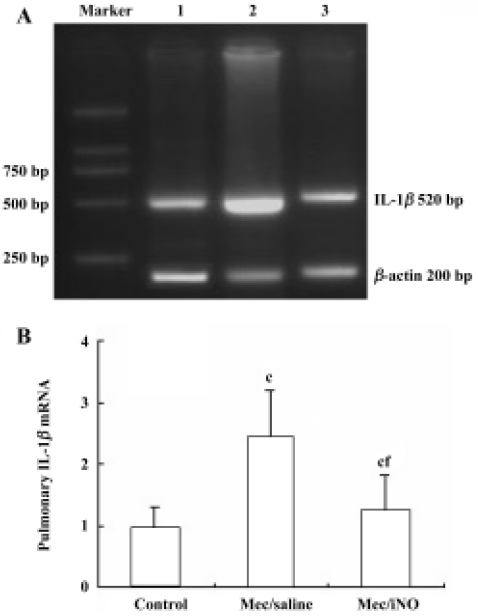
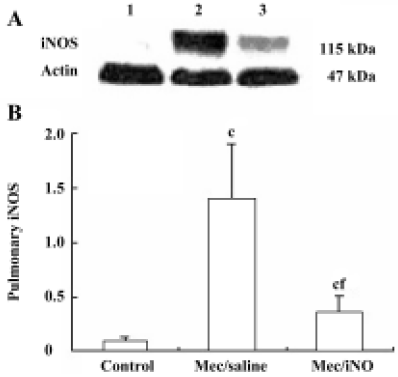
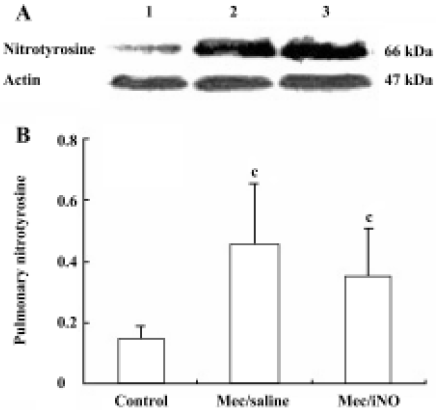
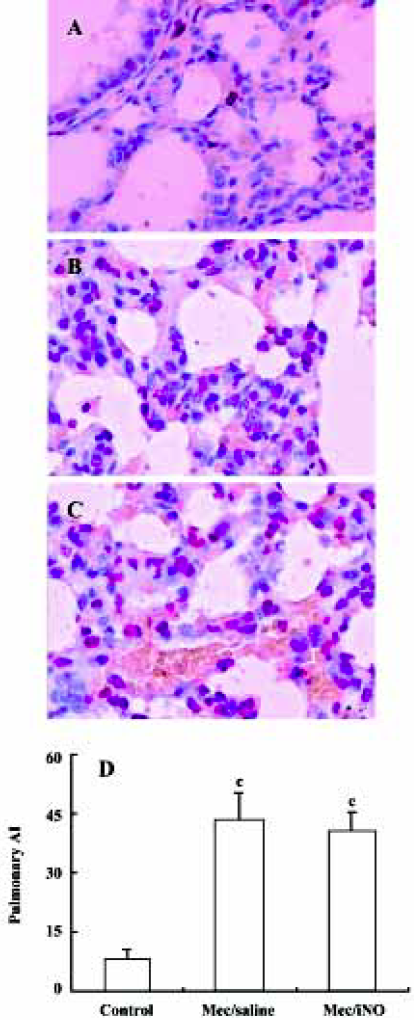
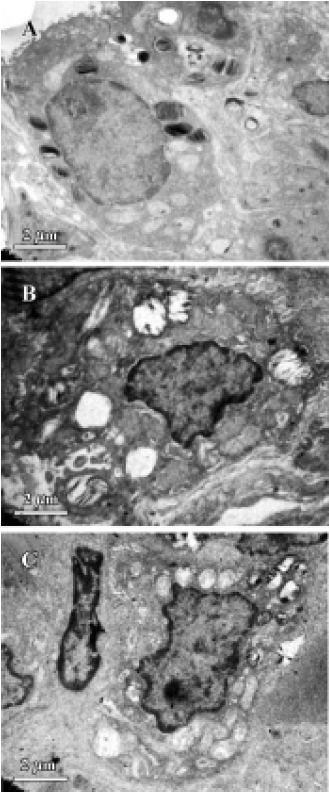
Inhalation of NO (20 μL/L) significantly inhibited meconium-induced iNOS protein and IL-1β mRNA expression when compared with the Mec/saline group (both P<0.01; Figures 1, 2). BAL cell count, MPO activity, and lung injury score were also decreased (P<0.01, P<0.01, and P<0.05, respectively; Table 1). However, there were no statistical differences in MDA level, nitrotyrosine formation or pulmonary AI between the Mec/saline and Mec/iNO groups (Table 1; Figures 3, 4). Electron microscopy revealed a significant amount of epithelial apoptosis in rat lung from both the Mec/saline and the Mec/iNO group (Figure 5).
Discussion
The present study shows that meconium aspiration in meconium-induced ALI, compared with NO inhalation, is associated with increased MPO activity, lung injury score, high numbers of neutrophils in BAL, and increased iNOS protein and IL-1β mRNA expression. It demonstrates that early NO inhalation at a dose of 20 μL/L. inhibits the pulmonary inflammatory injury, but does not increase pulmonary MDA and nitrotyrosine formation in meconium induced-ALI. These findings indicate that NO inhalation may protect the lung tissue against the meconium-induced pulmonary inflammatory response and that at the present dosage, the oxidative effects of iNO are not clinically significant.
Meconium contains variable amounts of proinflammatory substances, such as IL-1β, IL-6, IL-8, TNF-α, and heme, which may activate lung inflammatory cells to produce cytokines and elicit inflammatory reactions[11,14]. This is also demonstrated in the rat lungs in our study by induction of pulmonary IL-1β mRNA expression. As previously reported, high expression of iNOS might lead to overproduction of NO[15,16]. NO is an important mediator in inflammation and high levels of NO produced by iNOS can mediate lung injury[8]. During inflammatory conditions, meconium is able to activate macrophages to produce oxygen radicals, including superoxide anions[4]. Reaction of NO with superoxide anions produces peroxynitrite, which is a highly oxidative species and is capable of nitrating tyrosine residues of numerous proteins, leading to the formation of nitrotyrosine that may result in protein inactivation and lipid and DNA degradation. Nitrotyrosine is a product of the peroxynitrite pathway that has been used as a probe to detect NO-mediated oxidative reactions[8]. Our findings show that meconium aspiration results in the expression of iNOS and the production of MDA and nitrotyrosine, indicating that meconium-induced ALI is associated with pulmonary inflammatory and oxidative/nitrosative injury.
Findings from earlier studies suggest that inhaled NO can decrease inflammatory reactions in neonatal and adult lungs[6,17]. In the present study, we showed an attenuation in pulmonary inflammatory injury after NO treatment, similar to earlier findings. Evidence from our study shows that the mechanism of the anti-inflammatory effects of NO inhalation may be associated with inhibited expression of pulmonary iNOS protein and IL-1β mRNA. On the other hand, during inflammatory conditions, inhaled NO might increase the reaction between superoxide and NO to enhance tissue nitration and exacerbate injury. Nitrotyrosine has been found in the lungs of patients with acute respiratory distress syndrome who have received inhaled nitric oxide[18,19]. In newborn piglets the formation of peroxynitrite may be enhanced in lungs exposed to high doses of inhaled NO or hyperoxia[20]. However, inhaled NO might have contradictory effects on lung tissue: first, an increase in nitration as a result of increased amounts of NO and nitrite, thereby enhancing the production of peroxynitrite and the reaction via peroxidase as a source of nitrite[19]; and second, a decrease in nitration by inhibiting leukocyte accumulation in the lungs, thereby reducing the production of NO and superoxide[9,21]. In the present study, inhaled NO at 20 μL/L. does not further increase pulmonary nitrotyrosine formation. Some studies have also shown that early inhalation of NO in severe respiratory failure is not associated with increased NO toxicity. In the rat model of lipopolysaccharide-induced ALI, NO inhalation during the early phase of inflammation did not increase, but rather decreased tyrosine nitration and chlorination, possibly by reducing neutrophil sequestration[9]. Inhaled NO at 100 μL/L was shown to increase the survival of rats with hyperoxia[22]. Inhaled NO at 20 μL/L with concurrent hyperoxia did not increase intracellular or intercellular nitrotyrosine in a mouse model[23]. According to evidence from this and from previous studies, the oxidative effects of iNO are not clinically significant.
There is some indirect evidence suggesting that apopto-sis may play an important role in ALI. Aspirated meconium may cause apoptotic cell death of the airway epithelium, supposedly through the action of bile salt or pulmonary inflammatory cells. Increased release of reactive oxygen and nitrogen species by pulmonary inflammatory cells may be associated with apoptotic cell death in the lung. Previous studies indicate that NO may function as an inhibitor of the apoptotic death program. An earlier study showed that NO inhalation inhibited pulmonary apoptosis in porcine meconium aspiration[3]. However, there was a bulk of evidence indicating that NO induced apoptosis in different cell lines[7]. In this study we found that NO inhalation did not protect the lung from meconium-induced apoptosis, despite the positive effect on pulmonary inflammatory injury.
In summary, meconium-induced ALI is associated with inflammatory oxidant injury and epithelial apoptosis. Early inhalation of NO at a dose of 20 μL/L. does not seem to prevent epithelial apoptosis caused by meconium aspiration. Inhalation of NO at the present dose is not associated with any detectable increase in oxidative or nitrosative damage. The results presented here suggest a lack of toxicity of inhaled NO due to oxidant injury and further justify therapeutic trials on inhaled NO in clinical respiratory failure. Caution with regard to pulmonary toxicity is still required.
Acknowledgement
We thank Mr Zhong-sheng YU and Mr Yao-sheng DONG for excellent technical assistance.
References
- Cleary GM, Wiswell TE. Meconium-stained amniotic fluid and the meconium aspiration syndrome: an update. Pediatr Clin North Am 1998;45:511-29.
- MH, Bae CW. Inhibitory effect of meconium on pulmonary surfactant function tested in vitro using the stable microbubble test. Eur J Pediatr 2000;159:770-4.
- Holopainen R, Aho H, Laine J, Halkola L, Kaapa P. Nitric oxide inhalation inhibits apoptosis but not inflammatory injury in porcine meconium aspiration. Acta Pediatr 1999;8:1147-55.
- Soukka HR, Ahotupa M, Ruutu M, Kaapa PO. Meconium stimulates neutrophil oxidative burst. Am J Perinatol 2002;19:279-84.
- Miao CH, Sun B, Jiang H, Xue ZG, Lindwall R. Pharmacodynamics and pharmacokinetics of inhaled nitric oxide in dogs with septic acute respiratory distress syndrome. Acta Pharmacol Sin 2002;23:278-84.
- Kinsella JP, Parker TA, Galan H, Sheridan BC, Halbower AC, Abman SH. Effects of inhaled nitric oxide on pulmonary edema and lung neutrophil accumulation in severe experimental hyaline membrane disease. Pediatr Res 1997;41 Suppl:457-63.
- Brune B, von Knethen A, Sandau KB. Nitric oxide and its role in apoptosis. Eur J Pharmacol 1998;351:261-72.
- Lang JD, McArdle PJ, O’Reilly PJ, Matalon S. Oxidant-antioxidant balance in acute lung injury. Chest 2002;122 Suppl:S314-20.
- Hataishi R, Kobayashi H, Takahashi Y, Hirano S, Zapol WM, Jones RC. Myeloperoxidase-associated tyrosine nitration after intratracheal administration of lipopolysaccharide in rats. Anesthesiology 2002;97:887-95.
- Lu MP, Du LZ, Yu ZZ, Chen XX. Protective effects and mechanisms of recombinant human superoxide dismutase in acute lung injury of rats following meconium aspiration. Chin J Pediatr 2004;42:777-81.
- Zagariya A, Bhat R, Uhal B, Navale S, Freidine M, Vidyasagar D. Cell death and lung cell histology in meconium aspirated newborn rabbit lung. Eur J Pediatr 2000;159:819-26.
- Bradford M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of dye-binding. Anal Biochem 1976;72:248-54.
- Li HP, Li X, He GJ, Yi XH, Kaplan AP. The influence of dexamethasone on the proliferation and apoptosis of pulmonary inflammatory cells in bleomycin-induced pulmonary fibrosis in rats. Respirology 2004;9:25-32.
- de Beaufort AJ, Bakker AC, van Tol MJ, Poorthuis BJ, Schrama AJ, Berger HM. Meconium is a source of pro-inflammatory substances and can induce cytokine production in cultured A549 epithelial cells. Pediatr Res 2003;54:491-5.
- Ding HL, Zhu HF, Dong JW, Zhu WZ, Yang WW, Yang HT, et al. Inducible nitric oxide synthase contributes to intermittent hypoxia against ischemia/reperfusion injury. Acta Pharmacol Sin 2005;26:315-22.
- Li YH, Yan ZQ, Braune A, Tullus K. Meconium induces expression of inducible NO synthase and activation of NF-kappaB in rat alveolar macrophages. Pediatr Res 2001;49:820-5.
- Du LZ, Long XY. Effects of inhaled nitric oxide on lung injury and neutrophil accumulation in rabbits with meconium aspiration. Acta Pharmacol Sin 2002;23 Suppl:165-6.
- Lamb NJ, Quinlan GJ, Westerman ST, Gutteridge JM, Evans TW. Nitration of proteins in bronchoalveolar lavage fluid from patients with acute respiratory distress syndrome receiving inhaled nitric oxide. Am J Respir Crit Care Med 1999;160:1031-4.
- Weinberger B, Laskin DL, Heck DE, Laskin JD. The toxicology of inhaled nitric oxide. Toxicol Sci 2001;59:5-16.
- Youssef JA, Thibeault DW, Rezaiekhaligh MH, Mabry SM, Norberg MI, Truog WE. Influence of inhaled nitric oxide and hyperoxia on Na, K-ATPase expression and lung edema in newborn piglets. Biol Neonate 1999;75:199-209.
- Honda K, Kobayashi H, Hataishi R, Hirano S, Fukuyama N, Nakazawa H, et al. Inhaled nitric oxide reduces tyrosine nitration after lipopolysaccharide instillation into lungs of rats. Am J Respir Crit Care Med 1999;160:678-88.
- Sato Y, Walley KR, Klut ME, English D, D’yachkova Y, Hogg JC, et al. Nitric oxide reduces the sequestration of polymorphonuclear leukocytes in lung by changing deformability and CD18 expression. Am J Respir Crit Care Med 1999;159:1469-76.
- Lorch SA, Foust R 3rd, Gow A, Arkovitz M, Salzman AL, Szabo C, et al. Immunohistochemical localization of protein 3-nitrotyrosine and S-nitrosocysteine in a murine model of inhaled nitric oxide therapy. Pediatr Res 2000;47:798-805.