
Overexpression of heat-shock protein 20 in rat heart myogenic cells confers protection against simulated ischemia/reperfusion injury1
Introduction
Heat shock proteins (HSP) are highly conserved molecules that fulfill a range of functions, including cytoprotection and the intracellular assembly, folding, and translocation of oligomeric proteins[1]. Expression of these proteins can be induced by a range of cellular insults, which include high temperature, oxidative stress, viral infection, and nutritional deprivation[2].
Previous studies have demonstrated that increased expression of various HSP, such as HSP70, HSP60, HSP10, and HSP90, protected against stress insult. Recently, the cytoprotective properties of small HSP have drawn increased attention. Small HSP, including HSP20, HSP25, HSP27, αB-crystallin, and myotonic dystrophy kinase binding protein, are a group of proteins expressed in muscle tissues and share sequence homology of approximately 80–100 amino acids at the C terminus, known as the α crystallin domain[3,4]. It was reported that overexpression of HSP27 by transfection into rodent and Chinese hamster cell lines directly correlated with survival from hyperthermia[5–7]. Overexpression of αB-crystallin has a similar effect[8–10]. It was also reported that overexpression of both HSP27 and αB-crystallin protected against ischemic injury in cardiac myocytes[11].
HSP20 is a newly discovered small HSP that was co-purified with αB-crystallin and HSP27 from skeletal muscle by affinity chromatography[12]. Previous reports demonstrated that HSP20 redistributed from the cytosol to insoluble fractions and dissociated from the aggregated form to the small form when rat diaphragm was exposed to heat stress in vitro[13]. Stable overexpression of HSP20 in Chinese hamster ovary cells results in enhanced survival after heat shock, which is similar to the results for αB-crystallin[14]. However, the effect of HSP20 on ischemia-mediated injury in cardiac myocytes has not been explored.
In our previous study, we showed that overexpression of HSP20 in rat heart in vivo protected against simulated ischemia/reperfusion (SI/R) injury, the mechanism of which related to the reduction of necrosis and apoptosis of ventricular cardiomyocytes (unpublished data). In the present study, our aim was to determine whether increased expression of HSP20 exerted a protective effect against SI/R injury in cardiac myocytes. We demonstrated, for the first time, that overexpression of HSP20 protected against ischemic damage in rat H9c2 cardiomyocytes, the protective effect of HSP20 in vitro being due primarily to reduced necrotic and apoptotic cell death, possibly via the protein kinase C (PKC)/mitochondrial K+ATP channel (mito K+ATP) pathway.
Materials and methods
Construction of recombinant adenovirus The recombinant adenovirus encoding HSP20 (Ad.HSP20) and adenovirus encoding GFP (Ad.GFP) were prepared as described previously[15]. Adenovirus was propagated in 293 cells and purified by 2 rounds of CsCl density ultracentrifugation (4 oC, 13000×g for 105 min and 16 h, respectively). Viral stocks were desalted through a PD-10 desalting column (Amersham Biosciences UK, Buckinghamshire, UK) into a Tris-buffered solution (10 mmol/L Tris, pH 8.0, 2 mmol/L MgCl2 and 4% sucrose)[16], plaque-titered, aliquoted, and stored at -80 oC with 4% sucrose until use.
Cell culture and simulated ischemia/reperfusion H9c2 cells were purchased from ATCC, the Global Bioresource Center (No CRl-1446; Hong Kong, China) and were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Cells were infected with Ad.HSP20 or Ad.GFP at a multiplicity of infection of 50 in no-serum DMEM when the confluence was 60%. After 2 h, the medium was removed and supplemented with DMEM containing 1% FBS for 24 h. Infected cells and the efficiency of infection were monitored by GFP expression with the use of fluorescent microscopy. After 24 h of infec-tion, cells underwent 8 h of simulated ischemia and 24 h of reperfusion. Simulated ischemia was achieved by placing cells in a hypotonic balanced salt solution consisting of 1.3 mmol/L CaCl2, 5 mmol/L KCl, 0.3 mmol/L KH2PO4, 0.5 mmol/L MgCl2, 0.4 mmol/L MgSO4, 69 mmol/L NaCl, 4 mmol/L NaHCO3, and 0.3 mmol/L Na2HPO4 without glucose or serum, and hypoxia was induced for 8 h at 37 oC[17]. Hypoxia was attained with an airtight jar from which the oxygen was exhausted through the oxygen-consuming GasPak System from BBL Microbiology Systems (Cockeysville, MD)[17]. At the end of the experiment, the dishes were removed from the chamber with the medium, and cells were reperfused in no-serum DMEM for 24 h. The control H9c2 cells were cultured in DMEM supplemented with 10% FBS, and cells were cultured in no-serum DMEM when the confluence was 60%. After 2 h, the medium was removed and cells were supplemented with DMEM containing 1% FBS. After 24 h, the medium was removed and cells were supplemented with a balanced salt solution at 37 oC for 8 h. The medium was then removed and the cells were cultured in no-serum DMEM for 24 h at 37 oC. The supernatant and cells were assayed separately for both lactate dehydrogenase (LDH) or caspase-3 activity.
Evaluation of necrosis and apoptosis by flow cyto- metry After SI/R, cells were harvested, washed, and double-stained using an annexin V-fluorescein-isothiocyanate (FITC) apoptosis detection kit. This kit is based on the observation that soon after initiating apoptosis most cell types translocate the membrane phospholipid phosphatidylserine from the inner face of the plasma membrane to the cell surface. Annexin V has a strong affinity for phosphatidylserine and therefore serves as a probe for detecting apoptosis. Cells that have lost membrane integrity will show red staining (propidium iodide) throughout the nucleus and therefore will be easily distinguishable from apoptotic cells. Samples were incubated for 15 min in the dark with annexin V and propidium iodide, and flow cytometry was carried out using a FACScan (Becton Dickinson, Heidelberg, Germany). Annexin V-FITC and propidium iodide-related fluorescence were recorded using FL1-H (525 nm) and FL2-H (575 nm) filters, respectively.
Lactate dehydrogenase (LDH) release assay Lactate dehydrogenase activity released into the medium or remaining in the cells was determined with the use of an LDH assay kit (Roche Diagnostics, Basel, Switzerland). The LDH activity is determined in an enzymatic test. In the first step, NAD+ is reduced to NADH/H+ by the LDH-catalyzed conversion of lactate to pyruvate. In the second step the catalyst transfers H/H+ from NADH/H+ to the tetrazolium salt INT, which is reduced to formazan, and the absorbance of the samples is measured at 490 nm using an enzyme-linked immunosorbent assay reader. Necrosis of cardiomyocytes was evaluated by the percentage of LDH release, which was calculated by the LDH activity in the medium divided by the total enzyme activity (medium and remaining activity in the cells) after reperfusion for 24 h.
Measurement of caspase-3 activity Cardiomyocytes were examined for apoptosis after simulated reperfusion for 24 h. Caspase-3 activity was determined by use of the caspase-3 Colorimetric Assay (R&D Systems, Minneapolis, MN, USA). Cells that were suspected to be or had been induced to undergo apoptosis were first lysed to collect their intracellular contents. The cell lysate was then tested for protease activity by the addition of a caspase-specific peptide that was conjugated to the color reporter molecule p-nitroanilide (p-NA). Cleavage of the peptide by caspase released the chromophore p-NA, which was quantitated spectrophotometrically at a wavelength of 405 nm.
Statistical analysis Data are expressed as mean±SD. Difference was analyzed for significance by one-way repeated-measures ANOVA and further analyzed with the use of the Newman-Keuls test for multiple comparisons between treatment groups. The results were considered significant at P<0.05.
Results
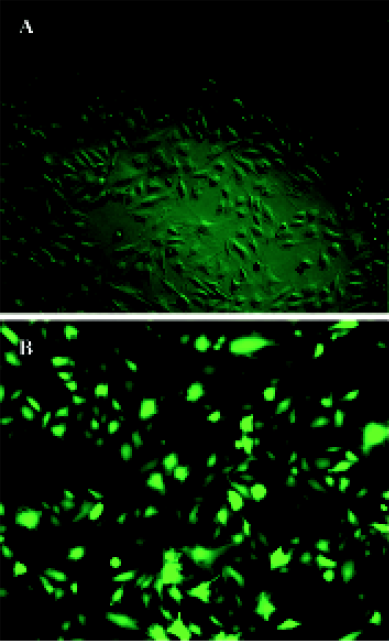
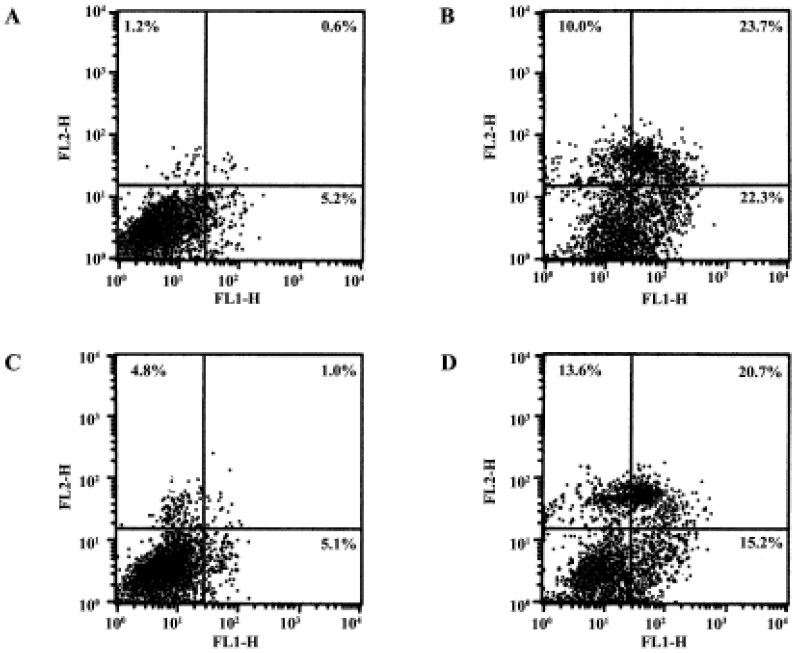
Gene transfer of Hsp20 inhibits necrosis and apoptosis of H9c2 cells We infected H9c2 cells with Ad.HSP20 or Ad.GFP. The infection efficiency was 94.7% ± 2.1% (n=5) with Ad.HSP20 and 95.2%±2.6% (n=5) with Ad.GFP, as indicated by GFP fluorescence (Figure 1). Importantly, there was no apparent morphological alteration or difference in the number of adherent cells between control cells, Ad.HSP20-infected cells, and Ad.GFP-infected cells. We determined the levels of necrosis and apoptosis of H9c2 cells double-stained by annexin V-FITC and propidium iodide after SI/R by flow cytometry. The lower-left quadrants of the cytograms showed the viable cells, which excluded propidium iodide-stained cells and cells that were negative for annexin V-FITC binding. The lower-right quadrants represented the apoptotic cells, annexin V-FITC-positive cells, and propidium iodide-negative cells. The upper-right quadrants contained necrotic and late-apoptotic cells, positive for annexin V binding and for propidium iodide uptake. The upper-left quadrants represented cells damaged during the procedure. For each treatment group, 10 000 cells were analyzed. In the control group, most of the cells were healthy (93.0%) (Figure 2A). Of the H9c2 cells exposed to SI/R, 22.3% were propidium iodide negative (lower-right quadrant) while 23.7% were propidium iodide positive (upper-right quadrant), which indicated early apoptosis and late apoptosis/necrosis, respectively (Figure 2B). Overexpression of HSP20 significantly reduced the number of cells labeled with annexin-V. The percentage of early apoptotic and late apoptotic/necrotic cells was significantly decreased to 5.1% and 1.0%, respectively (Figure 2C). H9c2 cells infected with control virus had no such result (Figure 2D).
We also detected necrosis and apoptosis by LDH release and caspase-3 activity, respectively. Cellular damage was measured by the amount of cytosolic LDH release after SI/R. Cell apoptosis was quantitated by caspase-3 activity. Overexpression of HSP20 in cardiomyocytes resulted in a 21.5% reduction in the proportion of cytosolic LDH release and a 58.8% reduction in caspase-3 activity, compared with Ad.GFP-treated cells (Figure 3B).
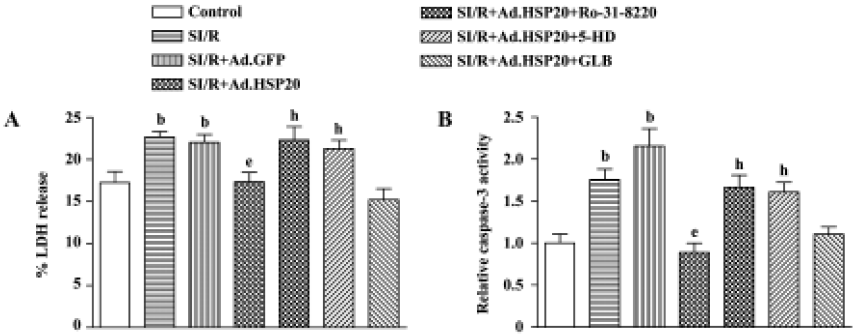
Protein kinase C inhibitor and mitochondrial K+ATP channel inhibitor reversed the protective effect of HSP20 Pretreatment with the PKC inhibitor Ro-31-8220 (0.1 µmol/L) for 30 min prior to SI/R reversed the protective effect of HSP20 on LDH release and caspase-3 activity (Figure 3). Pretreatment with the selective mitochondrial K+ATP channel inhibitor 5-hydroxydecanoate (5-HD, 100 µmol/L) for 30 min prior to SI/R also canceled the protective effect of HSP20, whereas pretreatment with the non-selective K+ATP channel inhibitor glibenclamide (100 µmol/L) had no significant effect (Figure 3). These data suggest that the protective effect of HSP20 in vitro is due to reduced necrotic and apoptotic death of cardiomyocytes, possibly via the PKC/mito K+ATP pathway.
Discussion
Our previous study demonstrated that overexpression of HSP20 in rat heart protected against ischemia/reperfusion, the protective effect relating to the reduction of necrosis and apoptosis of ventricular cardiomyocytes. In the present study, we used H9c2 cardiomyocytes, which originated from rat embryonic cardiac tissue and retained certain features of cardiac specificity[18,19], to investigate the mechanism of the protective effect of HSP20.
By detecting annexin V-FITC and propodium iodide double-stained H9c2 cells after SI/R by flow cytometry, we detected the necrosis and apoptosis of cells. We also detected the necrosis and apoptosis of H9c2 cells by LDH release and caspase-3 activity. Similar to the in vivo exp- eriments, our results in vitro suggest that the protective effect of HSP20 also relates to the reduction of necrosis and apoptosis, which is indicated by flow cytometry, LDH release and caspase-3 activity (unpublished data).
Myocardial ischemia is frequently followed by reperfusion. Reperfusion and the resultant reoxygenation lead to the generation of oxygen radicals that can cause reperfusion injury. In the present study, we used H9c2 to mimic in vivo ischemia/reperfusion injury. We used LDH to evaluate the cell damage in vitro. The overexpression of HSP20 protected H9c2 cells against SI/R injury by reducing the LDH level. These data indicate that HSP20 reduces necrosis of cardiomyocytes. Our in vitro results also showed that H9c2 cells with overexpressed HSP20 had reduced caspase-3 activity. These data indicate that the anti-apoptotic effect of HSP20 in cardiomyocytes is mediated through reduced caspase-3 activity. In addition, Fan et al[15] showed that the protective effects of HSP20 were further increased by the constitutively phosphorylated HSP20 mutant (S16D). In our study, the PKC inhibitor Ro-31-8220 (0.1 µmol/L) and selective mitochondrial K+ATP channel inhibitor 5-HD (100 µmol/L) blocked the HSP20-induced protective effect. Our results are similar to those of previous reports showing that the PKC inhibitor blocks the cardio- protective effect of the mitochondrial K+ATP channel[20,21], which indicates that PKC lies upstream of the mitochondrial K+ATP channel.
Our results suggest that the protective effect of HSP20 is attributed to a reduction of necrosis and apoptosis in cardiomyocytes. Therefore, the cardioprotective effect of HSP20 in vitro might be mediated mainly by inhibiting cardiomyocyte necrotic and apoptotic cell death, possibly via the PKC/mito K+ATP pathway.
Acknowledgements
We are grateful to Prof Rui-ping XIAO (Laboratory of Cardiovascular Science, Gerontology Research Center, National Institutes of Health, Bethesda, MD, USA) for her advice during this research.
References
- Hightower LE. Heat shock, stress proteins, chaperones and proteotoxicity. Cell 1991;66:191-7.
- Welch WJ. How cells respond to stress. Sci Am 1993;268:56-64.
- Boelens WC, Croes Y, de Ruwe M, de Reu L, de Jong WW. Negative charges in the C-terminal domain stabilize the αB-crystallin complex. J Biol Chem 1998;273:28085-90.
- Suzuki A, Sugiyama Y, Hayashi Y, Nyu IN, Yoshida M, Nonaka I, et al. A novel member of the small heat shock protein family, binds and activates the myotonic dystrophy protein kinase. J Cell Biol 1998;140:1113-24.
- Landry J, Chretien P, Lambert H, Hickey E, Weber LA. Heat shock resistance conferred by expression of the human hsp27 gene in rodent cells. J Cell Biol 1989;109:7-15.
- Chretien P, Landry J. Enhanced constitutive expression of the 27-kDa heat shock proteins in heat-resistant variants from Chinese hamster cells. J Cell Physiol 1988;137:157-66.
- Lavoie JN, Gingras-Breton G, Tanguay RM, Landry J. Induction of Chinese hamster hsp27 gene expression in mouse cells confers resistance to heat shock. J Biol Chem 1993;268:3420-9.
- Aoyama A, Frohli E, Schafer R, Klemenz R. αB Crystallin expression in mouse NIH 3T3 fibroblasts: glucocorticoid responsiveness and involvement in thermal protection. Mol Cell Biol 1993;13:1824-35.
- Blackburn R, Galoforo S, Berns CM, Ireland M, Cho JM, Corry PM, et al. Thermal response in murine L929 cells lacking αB crystallin expression and αB crystallin expressing L929 transfectants. Mol Cell Biochem 1996;55:51-60.
- Iwaki A, Iwaki T, Tateishi J, Goldman JE. Sense and antisense modification of glial alpha B-crystallin production results in alterations of stress fiber formation and thermoresistance. J Cell Biol 1994;125:1385-93.
- Martin JL, Mestril R, Hilal-Dandan R, Brunton LL, Dillmann WH. Small heat shock proteins and protection against ischemic injury in cardiac myocytes. Circulation 1997;96:4343-8.
- Kato K, Shinohara H, Goto S, Inaguma Y, Morishita R, Asan T. Copurification of small heat shock protein with alpha B crystallin from human skeletal muscle. J Biol Chem 1992;267:7718-25.
- Kato K, Goto S, Inaguma Y, Hasegawa K, Morishita R, Asano T. Purification and characterization of a 20-kDa protein that is highly homologous to αB crystallin. J Biol Chem 1994;269:15302-9.
- van de Klundert FAJM, van den Issel PRLA, Stege GJ, de Jong WW. Rat Hsp20 confers thermoresistance in a clonal survival assay, but fails to protect coexpressed luciferase in Chinese hamster ovary cells. Biochem Biophys Res Commun 1999;254:164-8.
- Fan GC, Chu G, Mitton B, Song Q, Yuan Q, Kranias EG. Small heat-shock protein Hsp20 phosphorylation inhibits β-agonist-induced cardiac apoptosis. Circ Res 2004;94:1474-82.
- Nyberg-Hoffman C, Aguilar-Cordova E. Instability of adenoviral vectors during transport and its implication for clinical studies. Nat Med 1999;5:955-7.
- Mestril R, Chi SH, Sayen MR, O’Reilly K, Dillmann WH. Expression of inducible stress protein 70 in rat heart myogenic cells confers protection against simulated ischemia-induced injury. J Clin Invest 1994;93:759-67.
- Heshscheler J, Meyer R, Plant S, Krautwurst D, Rosenthal W, Schultz G. Morphological, biochemical and electrophysiological characterization of a clonal cell (H9c2) line from rat heart. Circ Res 1991;69:1476-86.
- Kimes BW, Brandt BL. Properties of a clonal muscle cell line from rat heart. Exp Cell Res 1976;98:367-81.
- Wang Y, Ashraf M. Role of protein kinase C in mitochondrial KATP channel-mediated protection against Ca2+ overload injury in rat myocardium. Circ Res 1999;84:1156-65.
- Sato T, O’Rourke B, Marban E. Modulation of mitochondrial ATP dependent K+ channels by protein kinase C. Circ Res 1998;83:110-4.