
Histone deacetylase inhibitors for treatment of hepatocellular carcinoma
Introduction
Hepatocellular carcinoma (HCC) is currently the fifth most common solid tumor worldwide and the fourth most common cause of cancer-related death[1]. The incidence of HCC is approximately 1 000 000 cases worldwide; and although in Europe and the USA the incidence of HCC is 4/100 000, it is 120/100 000 in Asia and Sub-Saharan Africa[2]. In particular, statistical data from the last decade show that HCC is the second most prevalent cause of cancer deaths in men and the third most prevalent cause in women in mainland China, with more than 500 000 new cases every year[3,4]. Moreover, a rise in the incidence of disease and mortality from HCC, most likely reflecting the increased prevalence of hepatitis C virus (HCV), has recently been observed also in Japan, Western Europe and the USA[5,6].
The major etiologies of HCC are well defined, and some of the steps in its molecular pathogenesis have been elucidated. HCC arises commonly, but not exclusively, in the context of liver injury, which leads to inflammation, hepatocyte regeneration, liver matrix remodeling, fibrosis and, ultimately, cirrhosis. In fact, cirrhosis represents the most important risk factor for HCC (70%–90% of cases of HCC develop in cirrhotic liver) and it is principally imputable to chronic viral hepatitis B and/or C (HBV, HCV), although other risk factors are alcohol abuse, metabolic liver disease (such as hemochromatosis, α1-antitrypsin deficiency and steatosis), androgenic steroid use and aflatoxin exposure[7]. In particular, chronic HBV infection is strongly associated with hepatocellular cancer in China[8], while HCV infection and subsequently cirrhosis is the leading cause of chronic hepatitis and HCC in Japan and Western countries[9].
Current trends in the management of HCC
The rates of early detection, treatment and prevention of HCC are poor, and a majority of patients (70%–85%) are affected by advanced or unresectable disease. Despite the many treatment options, the prognosis of HCC remains dismal. In fact, even for those patients who undergo resection, the recurrence rate can be as high as 50% at 2 years[10,11] and a meta-analysis that evaluated the results of 37 randomized clinical trials of systemic and regional chemotherapy in more than 2000 HCC patients concluded that non-surgical therapies were ineffective or minimally effective[12]. In addition, most published studies of systemic chemotherapy report a response rate ranging from 0% to 25%; treatment failure is most likely due to the particular resistance to cytostatic agents displayed by HCC cells[12,13], which are known to express the multidrug-resistant gene MDR-1[14]. Liver-directed therapies, such as transarterial chemoembolization or percutaneous ethanol injection, are palliative treatments, with encouraging results only for patients with small HCC[15,16].
Alternative therapeutic approaches have been investigated, but with disappointing results. The effectiveness of hormone therapy with anti-estrogens, anti-androgens or somatostatin analogues has been studied in several trials[17,18], but these treatments were found to be generally ineffective or to not produce reproducible results[19].
At present, orthotopic liver transplantation is considered the only curative treatment option for HCC, bringing about an increase in the 5-year survival rate from the historical 20%–36% to the recent 61%, which is likely related to adoption of the Milan criteria at US transplantation centers[23,24]. However, most patients with cirrhosis are not generally considered good candidates for liver transplantation, or they remain on the waiting list until they die from tumor progression or cirrhosis-related complications.
An understanding of tumor biology and the key molecular events leading to HCC development is therefore fundamental for identifying new therapeutic strategies that are effective against HCC but not toxic to normal cells, and are well-tolerated by the typical patient with underlying cirrhosis.
HCC biology and targeted therapies
In most types of cancer, hepatocarcinogenesis is a multi-step process involving different genetic alterations, including cellular oncogene activation, tumor suppressor gene inactivation (possibly in cooperation with genomic instability), DNA repair defects, overexpression of growth and angiogenic factors, and telomerase activation, which ultimately lead to malignant transformation of the hepatocyte[20]. For a more comprehensive review of the complex molecular pathogenesis of HCC the reader may refer to some excellent papers including those by Ozturk[21], Moradpour and Wands[22], Thorgeirsson and Grisham[7] and Suriawinata and Xu[23].
On the basis of knowledge of the molecular pathways that are associated with the malignant phenotype, new and promising agents, which are specifically targeted to tumor cell receptors or signaling events, are emerging. This is the case for the selective growth factor receptors, tyrosine kinase inhibitors, the fundamental role of which is to block signal transduction[24,25], or farnesyltransferase inhibitors, which counteract oncogene ras-mediated signaling[26]. Moreover, to stimulate an immunological response against liver tumors, other agents have been tested, for example interferon-α, which can significantly prolong the survival of HCC patients when given at high doses[27], interleukin-2, which can produce objective remission when given alone[28] or in combination with melatonin[29], or tumor necrosis factor-α-related apoptosis-inducing ligand (TRAIL), which has not yet been tested in humans, but has been found to be effective in pre-clinical experiments[30].
Gene therapy is a new and promising therapeutic strategy that is based on the introduction of genetic material, for example natural genes, chimeric genes or subgenomic molecules, into cells in order to generate a beneficial effect against disease[31]. So far, a variety of gene therapy approaches have been designed to treat liver cancer, including the replacement of functional tumor suppressor genes[32], inhibition of oncogenes[33], selective prodrug activation within the tumor[34], stimulation of antitumor immunity[35] and inhibition of tumor vascularization[36], although encouraging results have been mostly only obtained in pre-clinical models.
Histone acetylation status and HCC develop-ment
In addition to the classical genetic alterations (chromo-somal deletions and rearrangements, and gene amplifications and mutations) first recognized as being responsible for hepatocarcinogenesis, molecular approaches have recently been used to identify alterations in the epigenetic control of gene transcription that positively mediate cellular proliferation or inactivate tumor suppressor genes[37]. In particular, acetylation, methylation and phosphorylation of the N-terminal lysine tails of the chromatine core histones have been found to play a critical role in post-translational modifications, suggesting the hypothesis of a histone “code”[38]. Among these modifications, the acetylation status of the histones constitutes the major epigenetic alteration, and is fundamentally involved in transcriptional regulation[39].
The acetylation level of histones is determined by the equilibrium between the activities of two groups of enzymes, histone acetyltransferases (HAT) and histone deacetylases (HDAC), which respectively add or remove acetyl groups from the lysine tails[40]. Hypoacetylated histones are associated with a more packaged chromatin structure and with suppression of gene transcription, whereas highly acetylated histones activate gene transcription by releasing the chromatin structure[41]. At present, eleven mammalian HDAC have been identified, and these have been ordered into 3 classes[42]. Class I deacetylases (HDAC 1, 2, 3, and 8) share homology in the catalytic sites; class II includes HDAC 4, 5, 6, 7, 9, 10, and 11, of which HDAC 4, 5, 7, and 9 share homology in the C-terminal catalytic domain and N-terminal regulatory domain, whereas HDAC11 contains conserved residues in the catalytic core regions shared by both classes I and II, and HDAC6 and HDAC10 have two regions that are homologous with the class II catalytic site. The third class of HDAC is the conserved nicotinamide adenine dinucleotide-dependent Sir2 family. Increasing evidence indicates that HDAC are not redundant in function and distribution: class I HDAC are found exclusively in the nucleus, whereas class II HDAC shuttle between the nucleus and cytoplasm in response to certain cellular signals[43]. HDAC do not bind directly to DNA, but are recruited by protein complexes that can differ in their subunit composition.
The balance between HAT and HDAC activity in regulating DNA folding and gene transcription can be disrupted by HDAC inhibitors, which act by blocking HDAC enzymes; HDAC inhibition leads to lysine residue hyperacetylation and to DNA conformation changes (Figure 1). Inaccessible promoter regions thus become available targets for transcription factors, which activate the re-expression of several genes, including those involved in cell growth arrest, differentiation and apoptosis[44,45].
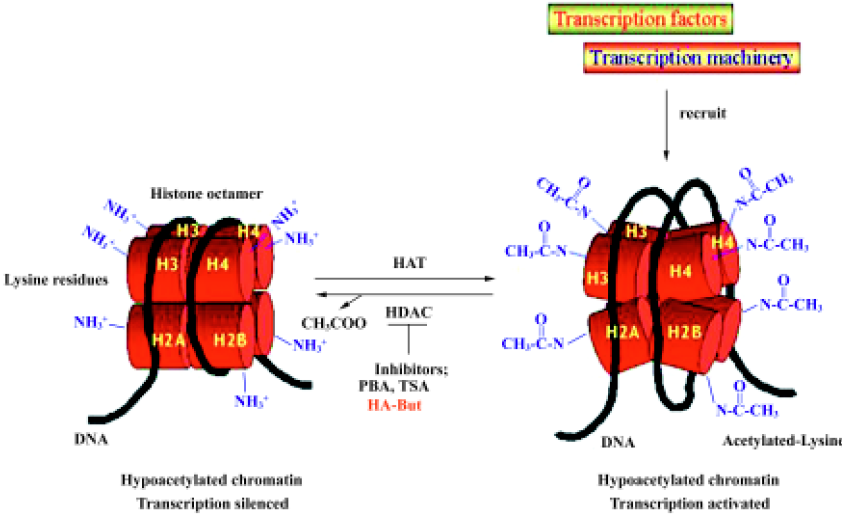
HDAC inhibitors and cancer treatment
Several lines of evidence suggest that inappropriate transcriptional activation commonly occurs in the formation of many types of cancer and that an imbalance between HAT and HDAC activity may be responsible for the alteration. Because HDAC dysregulation has been demonstrated in many solid cancers, including hepatocellular carcinoma, HDAC inhibitors have been investigated for their therapeutic potential to reprogram transcription and inhibit tumor cell growth and progression[46]. Historically, sodium butyrate (NB), which is normally present in the human colon as a product of the metabolic degradation of complex carbohydrates by colonic bacteria, was the first compound found to cause an increase in histone acetylation[47] and to regulate the physiological differentiation of colonocytes[48], suggesting its possible use in the prevention of colorectal cancer and the treatment of premalignant and neoplastic lesions. Many other HDAC inhibitors have since been identified, which belong to several chemical structure classes: 1) short-chain fatty acids, of which NB represents the prototype; 2) hydroxamic acids, including trichostatin A (TSA)[49] and a series of hydroxamic acid-based hybrid polar compounds, such as suberoylanilide hydroxamic acid (SAHA)[50]; 3) cyclic tetrapeptides, which may or may not contain the 2-amino-8-oxo-9,10 epoxy-decanoyl moiety (trapoxins A and B[51], FK228/depsipeptide[52] and apicidin[53]); and 4) benzamides (MS-275)[54] and others, such as valproic acid[55] and oxamflatin[56].
Several of these compounds, including SAHA, MS-275 and FK228/depsipeptide, are undergoing phase I and II clinical trials to examine their potential use as anticancer drugs for solid and hematological tumor treatment either as monotherapies or in combination with other cytotoxic and differentiation agents. For example, in a phase I clinical trial, SAHA was well tolerated when administered either intravenously or orally, and it was found to have antitumor activity in heavily pre-treated patients with advanced solid and hematological tumors. Furthermore, SAHA has good bioavaila-bility when administered orally and it induces objective responses in patients with prior therapy-resistant cutaneous T cell lymphomas[57]. Similarly, a phase II trial demonstrated that FK228/depsipepide was active against refractory neoplasms[58] and chronic lymphocytic and acute myeloid leukemia[59]. MS-275 is also well-tolerated when orally administered in patients with refractory or relapsed hematological malignancies, and it is biologically active in terms of histone acetylation[60]. However, none of these studies have been specifically focused on hepatocellular carcinoma, and in addition, despite the encouraging results obtained for several types of tumors, these drugs do not achieve the major goal in cancer therapy: to selectively target anti-cancer molecules to organs or compartments that harbor tumor cells.
HA-But as an HDAC inhibitor target delivery
As mentioned earlier in this paper, NB was the first compound found to cause an increase in histone acetylation[50], so given its antiproliferative and differentiation activities, together with a relative absence of systemic toxicity, it was a candidate for the prevention of colorectal cancer and a therapeutic agent for the treatment of pre-neoplastic and neoplastic lesions. Unfortunately, the first clinical study undertaken using high doses of NB resulted in only a partial and temporary remission, principally due to the relatively low potency of the drug and a low plasma concentration that was not sufficient to inhibit cell growth, but high enough to induce side-effects[61]. Both to overcome chemical constraints that restrict the clinical application of NB, and to specifically target the compound to cancer cells, a bioconjug-ate (HA-But), in which a hyaluronic acid backbone was used as a suitable carrier for butyric residues, was developed[62 ].
Hyaluronic acid (HA), also referred to as hyaluronan, is a polysaccharide molecule whose repeated disaccharide motif comprises D-glucuronic acid and D-N-acetylglucosamine linked together through alternating β-1,4 and β-1,3 glycosidic bonds. HA is present in all vertebrates and is a major constituent of the extracellular matrix, where it is organized, by specific interactions, with other matrix macromolecules[63]. HA has a high rate of turnover (in the bloodstream its half-life is 2–5 min) because it is rapidly captured by receptors on hepatic sinusoidal endothelial cells, which internalize it, and subsequently catabolize it in lysosomes. Sinusoidal endothelial cells actively remove almost 90% of the circulating HA, even though the spleen is also involved in its degradation[64]. The chemical properties of HA determine its physiological role as an essential structural element in the extracellular matrix, where it regulates the retention of water molecules in the interstitial space. In addition, HA provides support for cell orientation through some specific cell surface receptors, including CD44[65]. CD44 is a single-pass transmembrane glycoprotein consisting of 4 functional domains: the distal extracellular domain (responsible for the binding of HA), the membrane-proximal extracellular domain (whose sequence depends on the alternative CD44 mRNA splicing), the transmembrane domain (similar to that of many other single-pass proteins), and the cytoplasmic domain (which has protein motifs that either interact with the cytoskeletal proteins or are responsible for intracellular signaling)[68]. Although physiologically expressed by some normal human epithelial and mesenchymal cells, where it plays an important role in immune recognition, cell-cell aggregation and cell-matrix-cell signaling, CD44 is over-expressed in most human cancers, including hepatic carci-noma, and is associated with tumor progression[66,67]. In fact, clinical evidence indicates that in comparison with normal hepatocytes, HCC is frequently associated with an increased expression of CD44 receptors[73,74], an overexpression that provides them with an essential migration-promoting advantage as demonstrated by Lara-Pezzi et al[68]. In their interesting pre-clinical study, these authors demonstrated a link between CD44 expression and HBV infection. In fact, a CD44-dependent migratory phenotype was induced by the stable transfection of Chang liver cells with the gene coding for the hepatitis B virus X protein (HBx), which was able to enhance cell motility by altering the cellular morphology and inducing the formation of pseudopodal protrusions and cytoskeletal rearrangements together with the polarization of cell-surface adhesion molecule CD44. This finding is particularly relevant because it supports the relationship between HBV infection and hepatocarcinogenesis, underlining the role of HBx protein in the transformed phenotype.
In developing HA-But synthesis, we took advantage of some molecular properties of HA that satisfy some important biochemical concerns. In fact, HA can make stable bonds with butyric residues, increasing their in vivo half-life without affecting their pharmacological activity. In addition, we exploited its high affinity with the CD44 receptor, which is generally overexpressed on tumor cell membranes, to selectively target the butyric residues directly to neoplastic lesions, with minimal effects on normal cells. In fact, in agreement with data in the literature[69], we found that in normal cells such as fibroblasts, which express a percentage of CD44-positive cells (as evaluated by flow cytometry) similar to that of tumor cells, HA-But had no effect, suggesting that it is only really effective in actively proliferating cells, such as tumor cells (Figure 2).
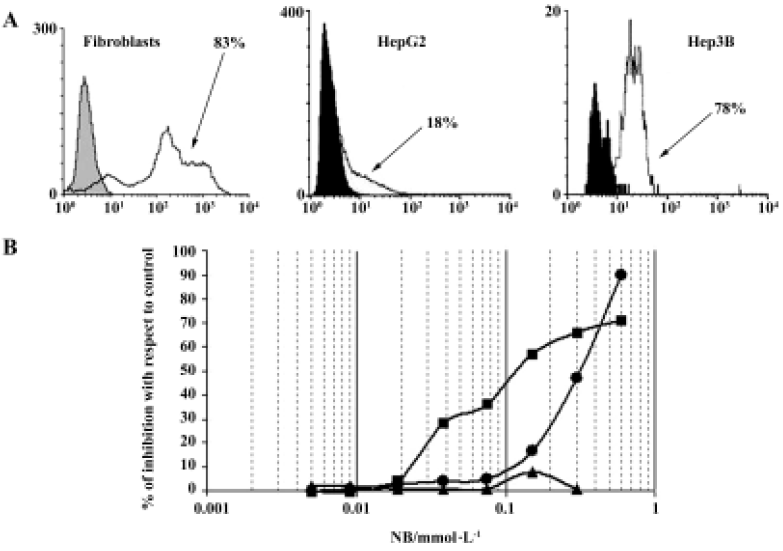
HA-But: pre-clinical in vitro results
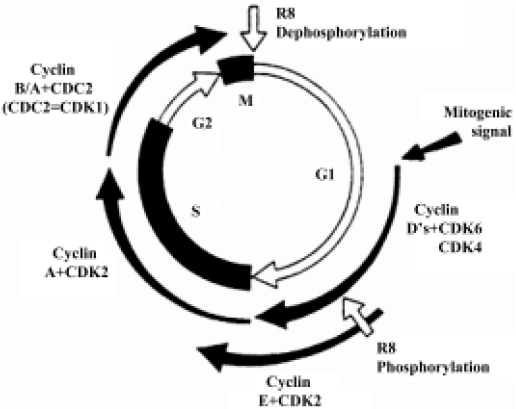
When we analyzed the antiproliferative effect of HA-But on two hepatocellular carcinoma cell lines, namely Hep3B and HepG2, which both expressed CD44 receptors (although to different extents), we found that after 6 days of treatment it exerted a dose-dependent effect, almost completely inhibiting the CD44-rich Hep3B (90%) cells, and moderately (but significantly) inhibiting (60%) the CD44-poor HepG2 cells; furthermore, this growth arrest corresponded to a block in the G0/G1 phase of the cell cycle[70]. As shown in Figure 3, the progression of the cell cycle is regulated by cyclin/cyclin-dependent kinase (cdk) complexes and cdk inhibitors, such as p16ink4, p21waf1, and p27kip1 proteins. Because it is known that overexpression of cdk inhibitors leads to cell-cycle arrest and apoptosis, we investigated the effect of HA-But on the expression of some of the molecules responsible for growth arrest, including cyclin D1, which is involved in the regulation of the G1 phase of the cell cycle, and whose gene is found amplified in 10%–20% of HCC,[71] but leads, when combined for example with p16ink4 amplification, to a loss of growth control in more than 30% of HCC. Experimental findings indicated that as in other types of tumors, such as lung cancer[65], and in agreement with the effect of other HDAC inhibitors on hepatoma cells[72,73], HA-But increased cdk-inhibitor expression (ie, the protein level of p27kip1 and p21waf1), while decreasing cyclin D1 protein levels, suggesting that the HA backbone does not interfere with the activity of butyric residues, which maintain their biological properties. In fact, as previously demonstrated in lung cancer[65], and similarly to NB, HA-But induces a hyperacetylation of histone H4, a dose-dependent overexpression of p27kip1 and p21waf1, and a block of cell growth in the G0/G1 phase of the cell cycle. In addition, cytometric analysis showed that CD44 receptor turnover was not affected by treatment with HA-But, which is a finding of great pharmacological relevance, because the stable presence of the receptors on plasma membranes guarantees a continuous internalization of the drug.
cDNA microarray analysis is a technical approach that enables investigators to measure the expression of thousands of mRNAs simultaneously in a biological specimen, providing comprehensive information that is useful for diagnosis and therapeutic intervention. Data gathered using this technique have further confirmed the previous experimental findings regarding the mechanism of action of HDAC inhibitors, and demonstrated that almost all HDAC inhibitors exerted their antiproliferative effects by modulating a small set of genes that regulate key cellular activities such as proliferation and differentiation. In particular, it has been shown that TSA, SAHA, depsipeptide are able to modulate genes involved in apoptosis and cell cycle pathways, among which are cdk inhibitors such as p16ink4, p21waf1, and bcl-2[74,75]. In addition, gene expression profiling of hepatocellular carcinomas has provided qualitative and quantitative evidence that the genes involved in the development and progression of a HCC correlate with the dysregulation of pathways associated with cell cycle regulation[76], apoptosis[77], signal transduction[78], cellular adhesion and angiogenesis[79,80]. Thus, the finding that HDAC inhibitors, including HA-But, can restore growth control and induce differentiation is particularly exciting.
HA-But: pre-clinical in vivo results
There are very few studies that have aimed to investigate the in vivo effect of HDAC inhibitors on tumor growth or the metastatic spread of liver tumors. Among the studies that have been conducted, a study using 4-phenylbutyrate, a derivative of the short-chain fatty acid butyrate, found that when administered via intratumor catheter, the compound reduced the growth of xenograft tumors derived from hepa-tocarcinoma cell lines, most likely by the induction of p21waf1 expression and the activation of apoptosis[78].
The in vivo capability of HA-But to inhibit primary tumor growth and metastatic spread has been investigated in several animal models[65,75], and evaluation of the drug biodistribution, by using the compound labeled with 99mTechnetium, found that a few minutes after iv admini-stration, there was a substantial accumulation of the compound in the liver, uniformly distributed in both lobes[75]. These results have been confirmed by the evaluation of the ex vivo distribution of HA-But, which showed that the liver was the organ of preferential accumulation, in agreement both with the finding obtained by using native HA[81] and with the observation that circulating hyaluronan is physiologically degraded by hepatic sinusoidal endothelial cells via CD44 receptors[82]. All these findings clearly suggest that iv injection can be appropriately exploited to treat intrahepatic lesions. In fact, when we explored the therapeutic potential of HA-But for the treatment of intrahepatic lesions induced in the mouse by intrasplenically inoculated Lewis lung carcinoma cells (LL3) and B16/F10 melanoma cells (two cell lines known for their particular aggressiveness[83,84], and which express high percentages of CD44-positive cells), we found that a prolonged iv administration of HA-But affected the survival time of tumor-bearing animals, reducing the number of intrahepatic metastatic lesions. In addition, prolonged treatment with low doses of HA-But significantly increased the survival time of treated mice relative to untreated controls. It is noteworthy that 90 d after tumor implantation, 80% of HA-But treated animals were still alive versus approximately one-third in the untreated group[75].
Conclusions and future perspectives
Experimental evidence and preliminary clinical phase I and II trials indicate that HDAC inhibitors acting on a pivotal mechanism of gene transcription such as histone acetylation may represent an innovative therapeutic approach for solid and hematological cancer. In particular, recent findings indicate that in pre-clinical studies HA-But is able to inhibit hepatocellular carcinoma cell growth, and that this antiproliferative activity is due to an increase in some cell-cycle related proteins, such as p21waf1 and p27kip1, and a decrease in some others, including cyclin D1. Furthermore, the results provide experimental evidence for the clinical use of HA-But as a promising agent for the treatment of hepatocellular carcinoma, a tumor that is otherwise particularly resistant to chemotherapy. The treatment exploits the overexpres-sion of CD44 receptors on tumor cell membranes, which allows selective targeting of the compound to the neoplastic lesion.
In addition to DNA acetylation, which hampers gene transcription, aberrant DNA hypermethylation of cytosine residues in the promoter region can cause growth-controlling gene silencing, a frequently observed phenomenon for several genes in HCC[7]. Therefore, the additional inhibition of DNA methyltransferase (DNMT), the enzyme responsible for such hypermethylation (the activity of which can be reversed using specific DNMT inhibitors, such as 5-aza-2-deoxycytidine, 5-AZA) could be an interesting candidate for further study. Therefore, a promising development for HA-But could be a new chemical compound in which HA-But is simultaneously esterified with 5-AZA. Such a molecule should be able to reactivate silenced genes and enhance the re-expression of specific genes involved in cell growth arrest, terminal differentiation and apoptosis in aggressive tumors such as hepatocellular carcinoma.
Acknowledgements
The authors wish to thank Drs I Scarlata, C Pellizzaro, and S Zorzet, who contributed to the HA-But synthesis, in vitro, and in vivo experiments, respectively.
References
- Parkin DM, Pisani P, Ferlay J. Estimates of the worldwide incidence of 25 major cancers in 1990. Int J Cancer 1999;80:827-41.
- Yu MC, Yuan JM, Govindarajan S, Ross RK. Epidemiology of hepatocellular carcinoma. Can J Gastroenterol 2000;14:703-9.
- Tang ZY. Hepatocellular carcinoma: cause, treatment and metastasis. World J Gastroenterol 2001;7:445-54.
- Parkin DM, Bray FI, Devesa SS. Cancer burden in the year 2000: The global picture. Eur J Cancer 2001;37:S4-66.
- Deuffic S, Poynard T, Buffat L, Valleron AJ. Trends in primary liver cancer. Lancet 1998;351:214-5.
- El-Serag HB, Devila JA, Petersen NJ, McGlynn KA. The continuing increase in the incidence of hepatocellular carcinoma in the United States: an update. Ann Intern Med 2003;139:817-23.
- Thorgeisson SS, Grisham JW. Molecular pathogenesis of human hepatocellular carcinoma. Nat Genet 2002;31:339-46.
- Ding X, Park YN, Taltavull TC, Thung SN, Jin XM, Jin Y, et al. Geographic characterization of hepatitis virus infections, genotyping of hepatitis B virus, and p53 mutation in hepatocellular carcinoma analyzed by in situ detection of viral genomes from carcinoma tissues: a comparison among six different countries. Jpn J Infect Dis 2003;56:12-8.
- National Institutes of Health Consensus Development Conference Statement: Management of Hepatitis C: 2002. Hepatology 2002;36:S1-20.
- Yamamoto J, Kosuge T, Takayama T, Shimada K, Yamasaki S, Ozaki H, et al. Recurrence of hepatocellular carcinoma after surgery. Br J Surg 1996;83:1219-22.
- Figueras J, Jaurrieta E, Valls C, Ramos E. Resection or transplantation for hepatocellular carcinoma in cirrhotic patients: outcomes based on indicated treatment strategy. J Am Coll Surg 2000;190:580-7.
- Lai EC, Lo CM, Fan ST, Liu CL, Wong J. Postoperative adjuvant chemotherapy after curative resection of hepatocellular carcinoma: a randomized controlled trial. Arch Surg 1998;133:183-8.
- Yamamoto M, Arii S, Sugahara K, Tobe T. Adjuvant oral chemotherapy to prevent recurrence after curative resection for hepatocellular carcinoma. Br J Surg 1996;83:336-40.
- Kato A, Miyazaki M, Ambiru S, Yoshitomi H, Ito H, Nakagawa K, et al. Multidrug resistance gene (MDR-1) expression as a useful prognostic factor in patients with human hepatocellular carcinoma after surgical resection. J Surg Oncol 2001;78:110-5.
- Camma C, Schepis F, Orlando A, Albanese M, Shahied L, Trevisani F, et al. Transarterial chemoembolization for unresectable hepatocellular carcinoma: Meta-analysis of randomized controlled trials. Radiology 2002;224:109-14.
- Vilana R, Bruix J, Bru C, Ayuso C, Sole M, Rodes J. Tumor size determines the efficacy of percutaneous ethanol injection for the treatment of small hepatocellular carcinoma. Hepatology 1992;16:353-7.
- CLIP Group (Cancer of the Liver Italian Programme). Tamoxifen in treatment of hepatocellular carcinoma: a randomized controlled trial. Lancet 1998;352:17-20.
- Kouroumalis E, Skordilis P, Thermos K, Vasilaki A, Moschandrea J, Manousos ON. Treatment of hepatocellular carcinoma with octreotide: a randomized controlled study. Gut 1998;42:442-7.
- Pignata S, Daniele B, Gallo C, De Vivo R, Monfardini S, Perrone F. Endocrine treatment of hepatocellular carcinoma: Any evidence of benefit? Eur J Cancer 1998;34:25-32.
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000;100:50-70.
- Ozturk M. Genetic aspects of hepatocellular carcinogenesis. Semin Liver Dis 1999;19:235-42.
- Moradpour D, Wands JR. Molecular pathogenesis of hepatocellular carcinoma. In: Zakim D, Boyer TD (eds), Hepatology. A Textbook of liver disease. Philadelphia: WB Sauders; 2002. p 1333–54.
- Suriawinata A, Xu R. An update on the molecular genetics of hepatocellular carcinoma. Semin Liver Dis 2004;24:77-88.
- Matsuo M, Sakurai H, Saiki I. ZD1839, a selective epidermal growth factor receptor tyrosine kinase inhibitor, shows antimeta-static activity using hepatocellular carcinoma model. Mol Cancer Ther 2003;2:557-61.
- Midgley R, Kerr D. Bevacizumab: current status and future directions. Ann Oncol 2005;16:999-1004.
- Sebti SM, Adjei AA. Farnesyltransferase inhibitors. Semin Oncol 2004;31:28-39.
- Lai CL, Lau JY, Wu PC, Ngan H, Chung HT, Mitchell SJ, et al. Recombinant interferon-alpha in inoperable hepatocellular carcinoma: a randomized controlled trial. Hepatology 1993;17:389-94.
- Palmieri G, Montella L, Milo M, Fiore R, Biondi E, Bianco AR, et al. Ultra-low-dose interleukin-2 in unresectable hepatocellular carcinoma. Am J Clin Oncol 2002;25:224-6.
- Alleghi R, Dissoni P, Barni S, Ardizzola A, Tanchini G, Piperno A, et al. Low-dose interleukin-2 subcutaneous immunotherapy in association with the pineal hormone melatonin as a first-line therapy in locally advanced or metastatic hepatocellular carcinoma. Eur J Cancer 1994;30A:167-70.
- Pei Z, Chu L, Zou W, Zhang Z, Qiu S, Qi R, et al. An oncolytic adenoviral vector of Smac increases antitumor activity of TRAIL against HCC in human cells and in mice. Hepatology 2004;39:1371-81.
- Sangro B, Herraiz M, Prieto J. Gene therapy of neoplastic liver diseases. Int J Biochem Cell Biol 2003;35:135-48.
- Anderson SC, Johnson DE, Harris MP, Engler H, Hancock W, Huang WM, et al. p53 gene therapy in a rat model of hepatocellular carcinoma: intra-arterial delivery of a recombinant adenovirus. Clin Cancer Res 1998;4:1649-59.
- Nguyen TH, Loux N, Dagher I, Vons C, Carey K, Briand P, et al. Improved gene transfer selectivity to hepatocarcinoma cells by retrovirus vector displaying single-chain variable fragment antibody against c-Met. Cancer Gene Ther 2003;10:840-9.
- Bilbao R, Gerolami R, Bralet MP, Qian C, Tran PL, Tennant B, et al. Transduction efficacy, antitumoral effect, and toxicity of adenovirus-mediated herpes simplex virus thymidine kinase/ganciclovir therapy of hepatocellular carcinoma: the woodchuck animal model. Cancer Gene Ther 2000;7:657-62.
- Sangro B, Mazzolini G, Ruiz J, Herraiz M, Quiroga J, Herrero I, et al. Phase I trial of intratumoral injection of an adenovirus encoding interleukin 12 for advanced digestive tumors. J Clin Oncol 2004;22:1389-97.
- Schmitz V, Wang L, Barajas M, Peng D, Prieto J, Qian C. A novel strategy for the generation of angiostatic kringle regions from a precursor derived from plasminogen. Gene Ther 2002;9:1600-6.
- Kondoh N, Wakatsuki T, Hada A, Shuda M, Tanaka K, Arai M, et al. Genetic and epigenetic events in human hepatocarcinogenesis. Int J Oncol 2001;18:1271-8.
- Jenuwein T, Allis CD. Translating the histone code. Science 2001;293:1074-80.
- Grunstein M. Histone acetylation in chromatin structure and transcription. Nature 1997;389:349-52.
- Wade PA, Pruss D, Wolffe AP. Histone acetylation: chromatin in action. Trends Biochem Sci 1997;22:128-32.
- Hebbes TR, Thorne AW, Crane-Robinson C. A direct link between core histone acetylation and transcriptionally active chromatin. EMBO J 1988;7:1395-402.
- de Ruijter Aj, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases: characterisation of the classical HDAC family. Biochem J 2003;3707:37-49.
- Khochbin S, Verdel A, Lemercier C, Seigneurin-Berny D. Functional significance of histone deacetylase diversity. Curr Opin Genet 2001;11:162-6.
- Marks PA, Rifkind RA, Richon VM, Breslow R, Miller T, Kelley WK. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer 2001;1:194-202.
- Csordas A. The biological role of histone acetylation. Biochemistry 1990;332:353-7.
- Marks PA, Richon VM, Breslow R, Rifkind RA. Histone deacetylase inhibitors as new cancer drugs. Curr Opin Oncol 2001;13:477-83.
- Vidali G, Boffa LC, Bradbury EM, Allfrey VG. Butyrate suppression of histone deacetylation leads to accumulation of multi-acetylated forms of histones H3 and H4 and increased DNase I sensitivity of the associated DNA sequences. Proc Natl Acad Sci USA 1978;75:2239-43.
- McIntyre A, Gibson PR, Young GP. Butyrate production from dietary fibre and protection against large bowel cancer in a rat model. Gut 1993;34:386-91.
- Yoshida M, Kijima M, Akita M, Beppu T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J Biol Chem 1990;265:17174-9.
- Richon VM, Zhou X, Rifkind RA, Marks PA. Histone deacetylase inhibitors: development of suberoylanilide hydroxamic acid (SAHA) for the treatment of cancers. Blood Cells Mol Dis 2001;27:260-4.
- Kijima M, Yoshida M, Sugita K, Horinouchi S, Beppu T. Trapoxin, an antitumor cyclic tetrapeptide, is an irreversible inhibitor of mammalian histone deacetylase. J Biol Chem 1993; 268: 22 429–35.
- Nakajima H, Kim YB, Erano H, Yoshida M, Horinouchi S. FR901228 a potent antitumor antibiotic is a novel histone deacetylase inhibitor. Exp Cell Res 1998;241:126-33.
- Darkin-Rattray SJ, Gurnett AM, Myers RW, Dulski PM, Crumley TM, Allocco JJ, et al. Apicidin: a novel antiprotozoal agent that inhibits parasite histone deacetylase. Proc Natl Acad Sci USA 1996;93:13143-7.
- Saito A, Yamashita T, Mariko Y, Nosaka Y, Tsuchiya K, Ando T, et al. A synthetic inhibitor of histone deacetylase, MS-275, with marked in vivo antitumor activity against human tumors. Proc Natl Acad Sci USA 1999;96:4592-7.
- Gottlicher M, Minucci S, Zhu P, Kramer OH, Schimpf A, Giavara S, et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J 2001;20:6969-78.
- Kim YB, Lee KH, Sugita K, Yoshida M, Horinouchi S. Oxamflatin is a novel antitumor compound that inhibits mammalian histone deacetylase. Oncogene 1999;18:2461-70.
- Kelly WK, Richon VM, O’Connor O, Curley T, MacGregor-Curtelli B, Tong W, et al. Phase I clinical trial of histone deacetylase inhibitor: suberoylanilide hydroxamic acid (SAHA) administered intravenously. Clin Cancer Res 2003;9:3578-88.
- Vigushin DM. FR-901228 Fujisawa/National Cancer Institute. Curr Opin Investig Drugs 2002;3:1396-402.
- Byrd JC, Marcucci G, Parthun MR, Xiao JJ, Klisovic RB, Moran M, et al. A phase 1 and pharmacodynamic study of depsipeptide (FK228) in chronic lymphocytic leukemia and acute myeloid leukemia. Blood 2005;105:959-67.
- Gojo I, Karp JE, Mann D. Phase I study of histone deacetylase inhibitor (HDI)MS-275 in adults with refractory or relapsed haematologic malignancies. Proc Am Soc Hem 2002; abstract no 2198.
- Miller AA, Kurschel E, Osieka R, Schmidt CG. Clinical pharmacology of sodium butyrate in patients with acute leukemia. Eur J Cancer Clin Oncol 1987;23:1283-7.
- Coradini D, Pellizzaro C, Abolafio G, Bosco M, Scarlata I, Stucchi L, et al. Hyaluronic acid butyric esters as promising antineoplastic agents in human lung carcinoma: a preclinical study. Invest New Drugs 2004;22:207-17.
- Tammi MI, Day AJ, Turley EA. Hyaluronan and homeostasis: a balancing act. J Biol Chem 2001;277:4581-4.
- Fraser JR, Alcorn D, Laurent TC, Robinson AD, Ryan GB. Uptake of circulating hyaluronic acid by the rat liver. Cell Tissue Res 1985;242:505-10.
- Isacke CM, Yarwood H. The hyaluronan receptor CD44. J Biochem Cell Biol 2002;34:718-21.
- Seiter S, Arch R, Reber S, Komitowski D, Hofmann M, Ponta H, et al. Prevention of tumor metastasis formation by anti-variant CD44. J Exp Med 1993;177:443-55.
- Gunthert U, Hofmann M, Rudy W, Reber S, Zoller M, Haussmann I, et al. A new variant of glycoprotein CD44 confers metastatic potential to rat carcinoma cells. Cell 1991;65:13-24.
- Lara-Pezzi E, Serrador JM, Montoya MC, Zamora D, Yanez-Mo M, Carretero M, et al. The hepatitis B virus protein (HBx) induces a migratory phenotype in a CD44-dependent manner: possible role of HBx in invasion and metastasis. Hepatology 2001;33:1270-81.
- Byrd JC, Shinn C, Ravi R, Willis CR, Waselenko JK, Flinn IW, et al. Depsipeptide (FR901228): a novel therapeutic agent with selective in vitro activity against human B-cell chronic lymphocytic leukaemia cells. Blood 1999;94:1401-8.
- Coradini D, Zorzet S, Rossin R, Scarlata I, Pellizzaro C, Turrin C, et al. Inhibition of hepatocellular carcinomas in vitro and hepatic metastases in vivo in mice by the histone deacetylase inhibitor HA-But. Clin Cancer Res 2004;10:4822-30.
- Nishida N, Fukuda Y, Komeda T, Kita R, Sando T, Furukawa M, et al. Amplification and overexpression of the cyclin D1 gene in aggressive human hepatocellular carcinoma. Cancer Res 1994;54:3107-10.
- Herold C, Ganslmayer M, Ocker M, Hermann M, Geerts A, Hahn EG, et al. The histone-deacetylase inhibitor Trichostatin A blocks proliferation and triggers apoptotic programs in hepatoma cells. J Hepatol 2002;36:233-40.
- Svechnikova I, Gray SG, Kundrotiene J, Ponthan F, Kogner P, Ekstrom TJ. Apoptosis and tumor remission in liver tumor xenografts by 4-phenylbutyrate. Int J Oncol 2003;22:579-88.
- Chiba T, Yokosuka O, Arai M, Tada M, Fukai K, Imazeki F, et al. Identification of genes up-regulated by histone deacetylase inhibition with cDNA microarray and exploration of epigenetic alterations on hepatoma cells. J Hepatol 2004;41:436-45.
- Peart MJ, Smyth GK, van Laar RK, Bowtell DD, Richon VM, Marks PA, et al. Identification and functional significance of genes regulated by structurally different histone deacetylase inhibitors. Proc Natl Acad Sci USA 2005;102:3697-702.
- Xu XR, Huang J, Xu ZG, Qian BZ, Zhu ZD, Yan Q, et al. Insight into hepatocellular carcinogenesis at transcriptome level by comparing gene expression profiles of hepatocellular carcinoma with those of corresponding noncancerous liver. Proc Natl Acad Sci USA 2001;98:15089-94.
- Tannapfel A, Wittekind C. Genes involved in hepatocellular carcinoma: deregulation in cell cycling and apoptosis. Virchows Arch 2002;440:345-52.
- McGary CT, Yannariello-Brown J, Kim DW, Stinson TC, Weigel PH. Degradation and intracellular accumulation of a residualizing hyaluronan derivative by liver endothelial cells. Hepatology 1993;18:1465-76.
- Chen X, Cheung ST, So S, Fan ST, Barry C, Higgins J, et al. Gene expression patterns in human liver cancers. Mol Biol Cell 2002;13:1929-39.
- Delpuech O, Trabut JB, Carnot F, Feuillard J, Brechot C, Kremsdorf D. Identification, using cDNA microarray analysis, of distinct gene expression profiles associated with pathological and virological features of hepatocellular carcinoma. Oncogene 2002;21:2926-37.
- Gustafsson S, Bjorknan T, Westlin JE. Labelling of high molecular weight hyaluronan with 125-I-tyrosine. Studies in vitro and in vivo in the rat. Glyco Conj J 1994;11:608-13.
- Saegusa S, Shuji I, Kawarada Y. Changes in serum hyaluronic acid levels and expression of CD44 and CD44 mRNA in hepatic sinusoidal endothelial hepatectomy in cirrhotic rats. World J Surg 2002;26:694-9.
- Barbara-Guillem E, Alonso-Varona A, Vidal-Vanaclocha F. Selective implantation and growth in rats and mice of experimental liver metastasis in acinar zone one. Cancer Res 1989;49:4003-10.
- Carrascal MT, Mendoza L, Valcarcel M, Salado C, Egilegor E. Interleukin-18 binding protein reduces B16 melanoma hepatic metastasis in neutralizing adhesiveness and growth factors of sinusoidal endothelium. Cancer Res 2003;63:491-7.