
Oxidative stress and stress signaling: menace of diabetic cardiomyopathy
Introduction
Cardiovascular disease is the leading cause of death in Western societies, causing approximately one million deaths annually[1]. It is both a primary and a secondary disease, resulting from several other disorders, such as hypertension, diabetes, alcoholism, obesity and the metabolic syndrome. Cardiovascular disease is defined as any disorder that decreases normal heart and/or vascular function. One of the leading causes of cardiovascular disease is diabetes, of both type 1 and type 2 origins. The incidence of diabetes is predicted to double by the year 2030 because of people’s sedentary lifestyles and an ever-growing cluster of pre-diabetic syndromes including metabolic syndrome, obesity, and insulin resistance[2]. Almost all of these metabolic disturbances are considered major risk factors in the development of heart dysfunction and congestive heart failure[3–7]. Nevertheless, the cellular mechanisms that relate to the development of cardiovascular complications in patients with diabetes have not been fully elucidated.
Diabetes mellitus is a group of metabolic disorders that are characterized by hyperglycemia resulting from defects in insulin secretion, action or both. Chronic hyperglycemia causes end-stage organ damage, dysfunction and failure of various organs, including the kidneys, nerves, eyes, blood vessels and hearts. Several pathological processes are involved in the development of diabetes, ranging from autoimmune destruction of the β-cells of the pancreas with resultant insulin deficiency (type 1 diabetes) to abnormalities that result in resistance to insulin (type 2 diabetes). Impairment of insulin action and resistance to the action of insulin often coexist in the same patient, so it is unclear which abnormality is responsible for the hyperglycemia. Symptoms of chronic hyperglycemia include polyuria, polydipsia, weight loss and blurred vision. Long-term complications of diabetes include retinopathy[8] with potential loss of vision; peripheral neuropathy[9] with risk of amputation; autonomic neuropathy causing gastrointestinal, genitourinary and cardiovascular symptoms; and nephropathy[10] leading to kidney failure.
Functional alterations in diabetic cardiomyopathy
Sustained diabetes mellitus leads to a deterioration of heart function that is known as diabetic cardiomyopathy, which occurs independently of the macro- and micro-vascular diseases that are frequently seen in diabetic patients[11]. Diastolic dysfunction is the most prominent mechanical defect in diabetic cardiomyopathy and is characterized by decreased compliance and slower rates of myocardial relaxation[11–13]. Both systolic and diastolic dysfunctions have been characterized to include prolonged contraction and relaxation, reduced velocity of contraction and relaxation, and depressed myocardial contractility in whole heart, tissue, and isolated ventricular myocytes from both diabetic patients and experimental animals[11–13]. Functional changes have been observed by electrocardiogram and echo-cardiography, which are manifested by shorter left ventricular ejection time, increased pre-ejection period, increased wall stiffness, decreased fractional shortening, decreased rate of left ventricular filling and increased action potential duration in diabetes[14,15]. All of these findings suggest that diabetic cardiomyopathy is represented by left ventricular dysfunction.
The chronic alterations at the end stages of diabetes are believed to be due to increased glucose levels[16,17]. Although the pathogenesis of diabetic cardiomyopathy has not been precisely described, several mechanisms have been speculated, including reduced energy production because of decreases in mitochondrial respiration and pyruvate dehydrogenase activity, accumulation of free radical species, glucose toxicity-induced oxidative stress and malfunction of cardiac contractile and intracellular Ca2+ regulatory proteins such as myosin, sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA), and Na+-Ca2+ exchanger[18–21]. The increased risk of diabetic cardiomyopathy and other heart complications warrants stringent and aggressive treatment against hyperglycemia, hyperinsulinemia, dyslipidemia and oxidative stress.
The most commonly used therapeutic regimes in diabetic patients with heart dysfunction include angiotensin-converting enzyme inhibitors, digoxin, diuretics, β-blockers, Ca2+ antagonists and spironolactone. Insulin-sensitizing agents such as thiazolidinediones are often prescribed in the treatment of diabetes more often than insulin-secretion-enhancing agents to avoid hyperinsulinemia and insulin resistance. In addition to pharmacological interventions, primary care for diabetic patients also includes lifestyle modifications such as smoking cessation, weight control, exercise and dietary restriction[7].
Diabetic cardiomyopathy type 1 diabetes
Type 1 diabetes mellitus leads to a cardiomyopathy in both human and animal models. The existence of a diabetic cardiomyopathy in humans is based on the presentation of ventricular dysfunction in patients without evidence of any other known cardiovascular disease (reviewed in Sowers et al [22] and Spector[23]). The clinical presentation of diabetic cardiomyopathy in type 1 diabetic patients was first presented by Rubler et al[24] based on four diabetic patients who suffered from congestive heart failure (CHF) in the absence of discernable coronary artery disease, valvular or congenital heart disease, hypertension or alcoholism. Numerous studies carried out since 1972 have supported the view that diabetic cardiomyopathy is a pervasive problem in type 1 diabetes and is first manifested by diastolic dysfunction[11,23]. Diabetic cardiomyopathy in experimental animal models of type 1 diabetes is characterized by phenotypic changes in the ventricular myocytes that occur in the presence or absence of coronary artery disease. This cardiomyopathy is well described in animal models with long-term type 1 diabetes and results in abnormal cardiomyocyte excitation-contraction (E-C) coupling [eg, prolonged action potentials, slowed cytosolic Ca2+ effluxes and slowed myocyte shortening and relengthening (reviewed by Pierce and Russell[25] and Chatham et al[26])]. The cellular mechanisms that contribute to myocyte dysfunction involve depressed expression and function of SERCA and Na+/Ca2+ exchanger (NCX)[27]. Regulation of E-C coupling is also impaired in diabetic hearts, such that β-adrenergic receptor signaling is depressed, which may result from changes in β-adrenergic receptor density or redistribution of β-adrenergic receptor subtypes[28], or perhaps signaling downstream of the receptors[29]. Elevated protein kinase C (PKC) activity and changes in the expression of specific PKC isoforms are also found in type I diabetic hearts[30,31].
Diabetic cardiomyopathy in type 2 diabetes
The more prevalent type 2 diabetes is a combination of resistance to insulin action and an inadequate compensatory insulin secretory response. Type 2 diabetic patients are able to survive with little or no insulin supplementation; however dietary modification and exercise are imperative for living with the disorder. Diabetic cardiomyopathy has also been determined clinically in the type 2 diabetic patients. Investigators in the Strong Heart Study (SHS) performed detailed echocardiographic analysis of a population of American Indians with a high rate of type 2 diabetes and reported that diabetes was associated with increased left ventricular (LV) mass, LV wall thickness, reduced systolic and particularly diastolic function, independent of hypertension[32,33]. Similar to diabetes, investigators from the Framingham Heart Study reported increased LV mass and wall thickness in individuals with glucose intolerance and insulin resistance[34]. There was also an association between left atrial (LA) size and insulin resistance. Since no relationship was found between systolic function and insulin resistance, one explanation for the relationship between LA size and insulin resistance offered was that the rising LA size might reflect the presence of diastolic dysfunction in insulin resistance (type 2 diabetes)[34]. Other studies in patients with diagnosed type 2 diabetes support the notion that the earliest cardiac abnormality is diastolic dysfunction[35]. Several investigators have experimentally shown that diabetes mellitus is associated with a specific cardiomyopathy[12,13] and depressed cardiac function independent of macro-/micro-vascular disease, suggesting the existence of a primary myocardial defect in both type 1 and type 2 diabetes mellitus[13,36].
Impaired insulin action (ie, insulin resistance) is characterized by a compensatory hyperinsulinemia and hyperlipidemia, which are major metabolic dysfunctions associated with the early stages of type 2 diabetes. Elevated plasma insulin and lipid levels can lead to numerous metabolic and pathophysiological derangements in various tissues, including the heart. Abnormal ventricular systolic and diastolic functions are reported in type 2 patients presenting without macrovascular disease or hypertension, providing indirect evidence that there is a diabetic cardiomyopathy in humans[37]. Furthermore, there is a considerable evidence that diastolic dysfunction occurs early in the disease process, which may contribute to high cardiac mortality among diabetic patients[38,39]. In clinical studies, detectable cardiac dysfunction has been reported to occur as early as the glucose intolerance phase (ie, hyperinsulinemmia and hyperglycemia) that follows insulin resistance[40].
The risk of congestive heart failure and other cardiovascular diseases is greatly increased in diabetic patients[41]. The Framingham Heart Study revealed that diabetic men had more than twice the frequency of congestive heart failure than did non-diabetic males, whereas diabetic women had a risk that was five times greater than non-diabetic women[34]. The development of diabetic cardiomyopathy is dependent on many factors; however, it is likely that all patients with diabetes will eventually develop some degree of diabetic cardiomyopathy.
Mechanisms of diabetic cardiomyopathy
Diabetic cardiomyopathy is a major reason for the high morbidity and mortality in diabetic populations[41], particularly among elderly people and postmenopausal women. In adult patients with diabetes, the risk of cardiovascular disease is three to five times greater than that in the general population. Several mechanisms for the development of cardiomyopathy have been postulated, including alterations in intracellular ion homeostasis, reduction in intracellular energy metabolism, alteration in glucose metabolism, disrupted polyol pathway and enhanced oxidative stress. Several mechanisms have been proposed to explain how all of the pathologies involved in the progression of diabetic cardiomyopathy can result from hyperglycemia. Four main hypotheses have been presented to describe how hyperglycemia can cause all of these diabetic complications[42]: increased polyol pathway flux, increased advanced glycation end-product (AGE) formation, increased protein kinase C isoform expression, and increased hexosamine pathway flux (Figure 1). We hope to show that all of these pathways, as well as several others, lead to hyperglycemia and increased reactive oxygen species (ROS) formation, causing diabetic cardiomyopathy.
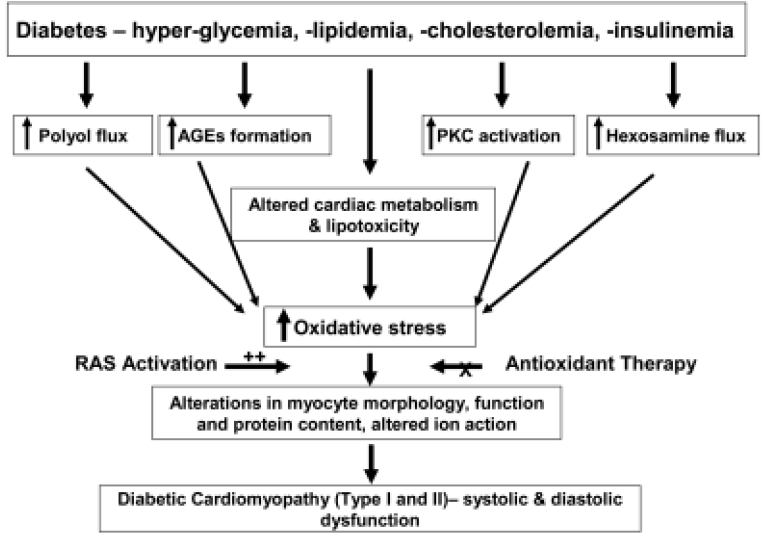
Alterations in intracellular ions
Changes in intracellular cations are directly related to the altered electromechanical activities of diabetic hearts. Alterations in intracellular Na+ are often accompanied by a decrease in K+ and Ca2+ in diabetic heart, which may be related in part to a diabetes-induced conformational change in the Na+-K+-ATPase pump[43]. Decreases in Ca2+ uptake, Ca2+ binding to the sarcolemma and Ca2+ intake by the myofibrillar Ca2+-ATPase activity have all been shown in diabetic rat hearts[43], which are partially reversible by insulin supple-mentation. Although the importance of these alterations in myocardial dysfunction seen in diabetes is not yet clear, the presence of abnormalities in Ca2+ handling and β-adrenergic stimulation is of paramount importance in understanding these myocardial dysfunctional changes[26]. Alterations in α1-adrenergic signaling in streptozotocin (STZ)-induced diabetic rat hearts have also been shown.
Increased polyol pathway flux
Increased glucose utilization by aldose reductase has been implicated in the development of diabetes complications. Aldose reductase is the first enzyme in the polyol pathway, which catalyses the NADPH-dependent reduction of carbonyl compounds, including glucose. In the polyol pathway, glucose is reduced to sorbitol by aldose reductase in the presence of NADPH, and sorbitol is then oxidized by sorbitol dehydrogenase (SDH) to fructose at the cost of NAD+. Normally, aldose reductase has a low affinity for glucose in the non-diabetic, low-glucose state. However the affinity for glucose is dramatically increased in the diabetic, high-glucose state, which converts glucose to sorbitol, with resultant decreases in NADPH. It has been demonstrated that increased aldose reductase activity in diabetic animals is correlated with increased NADH/NAD+[44]. There is further support from the facts that sorbitol and fructose levels in diabetic hearts are approximately nine-fold higher, and NADH/NAD+ levels in diabetic hearts are approximately four-fold higher than in normal hearts[44]. Experiments have demonstrated that cytosolic NADH/NAD+ was reduced by both SDH[45] and aldose reductase inhibition[44,46]. These data indicate that the polyol pathway is a target for cardioprotective interventions. This notion is supported by the observation that nitric oxide (NO) maintains aldose reductase in an inactive state and that this repression is relieved in diabetic tissues[47]. Thus, increasing NO availability may be a useful strategy for inhibiting the polyol pathway and preventing the development of diabetes complications. It is worth mentioning that contributions of the polyol pathway in diabetes are often tissue- and species-specific and may not fully explain the pathogenesis of all forms of diabetic complications[42].
One mechanism whereby increased polyol pathway flux leads to the complications of diabetes discussed earlier is that oxidation of sorbitol by NAD+ increases the cytosolic NADH:NAD+ ratio, leading to inhibition of the enzyme glyceraldehydes-3-phosphate dehydrogenase (GADPH) and increased concentrations of triose phosphate[42]. Increased triose phosphate could potentially increase the formation of AGEs and diacylglycerol (DAG), thus activating PKC isoforms[42].
Another mechanism whereby increased polyol pathway flux causes deleterious effects is that reduction of glucose to sorbitol by NADPH consumes NADPH, a cofactor in the generation of reduced glutathione (GSH), which could lead to increased oxidative stress. It has been shown that decreased levels of GSH are present in the lenses of transgenic mice overexpressing aldose reductase[48]. These observations have led many people to believe that this is the main mechanism whereby increased polyol pathway flux leads to many of the complications seen in diabetes.
Increased advanced glycation end products formation
Advanced glycation endproducts are enhanced in the presence of hyperglycemia and oxidative stress[49,50]. They bind to their cell-surface receptors (RAGE) resulting in the activation of postreceptor signaling, generation of intracellular ROS and the activation of gene expression[51–58]. AGEs have also been shown to be mediators of late diabetic complications and chronic vascular disease[59].
The importance of AGEs in the development of diabetic complications is seen in the observation that two structurally similar AGE inhibitors partially prevented diabetic complications in the retina, kidney and nervous system[60–62]. One of the mechanisms how AGE precursors target cells is through the binding of AGE receptors to endothelial cells, mesangial cells and macrophages, inducing receptor-mediated production of ROS. This receptor ligation increases the production of the transcription factor NF-κB, also causing increased oxidative stress.
Some of the most striking data on the role of AGE and the development of diabetic complications has been obtained when looking at the receptor for AGE, RAGE. RAGE suppressed the formation of macrovascular disease in a type I diabetic mouse model in a glucose- and lipid-independent fashion[63]. It has also been shown that blockage of RAGE inhibited the formation of diabetic nephropathy and enhanced wound repair (known to be a problem in diabetics) in murine models. The mechanism involved in RAGE-associated development of diabetic complications appears to be related to increased production of ROS[42].
Activation of protein kinase C isoforms
Protein kinase C is also increased in the tissues of diabetic patients[64]. Activation of the PKC pathway by hyperglycemia can occur directly or indirectly (via ligation of AGE receptors[65] or increased activity of the polyol pathway[66]) and can synergize with other kinase pathways, that is, the MAPK pathway. Interactions between these pathways are likely to play a role in determining the long-term effects of hyperglycemia. As discussed earlier, sorbitol, whose formation from glucose is catalyzed by aldose reductase, is increased when intracellular glucose concentrations rise[67], and can accumulate intracellularly, which can cause cell damage. The p38 mitogen-activated protein kinase (MAPK) and c-Jun N-terminal kinase (JNK) pathways are also activated by sorbitol. The significance of the sorbitol pathway as a cause of diabetic complications was demonstrated in transgenic mice that overexpressed the aldose reductase gene[48,68–70] and by data showing that inhibitors of this enzyme prevent the development of long-term diabetic complications in these animals[71].
Increased hexosamine flux and glucose auto-oxidation
Hyperglycemia increases flux through the hexosamine pathway by providing more fructose-6-phosphate for glutamine: fructose-6-phosphate amidotransferase (GFAT), the rate-limiting enzyme of the pathway. The effect of hyperglycemia on flux of the hexoasamine pathway probably reflects increased fructose-6-phosphate levels, which result from inhibition of GAPDH by ROS[72].
Recently, ROS formation due to glucose autooxidation has been hypothesized to play a role in the pathogenesis of diabetic cardiomyopathy in diabetic populations; however, no unifying hypothesis exists as to how glucose autooxidation causes any of the complications seen in diabetes.
Alterations in stress signaling pathways
Hyperglycemia in diabetes causes changes in membrane function and metabolic and biochemical alterations within days, changes in contractile function within weeks, and morphological changes and heart dysfunction within months[26]. A significant increase in oxidative damage via lipid peroxidation was observed in the hearts of diabetic rats[73]. Production of hydroxyl radicals was also detected in diabetic rats induced by streptozotocin[74]. In the heart, hydroxyl radical production and elevated blood glucose concentration were directly correlated with the amount of STZ injected into rats, up to 60 mg/kg body weight[74]. With the use of fluorescent probes, myocytes isolated from STZ-induced diabetic mice were used to detect hydrogen peroxide and hydroxyl radicals, and increased ROS was observed compared with control mice[21]. Oxidative damage caused by ROS has been shown to lead to multiple complications of diabetes[75–78]. Blocking ROS and superoxide formation, however, has been shown to prevent hyperglycemia-induced organ damage in diabetes[79].
One major intracellular target of hyperglycemia and oxidative stress is NF-κB[80–84], which can be activated by a variety of exogenous and endogenous stimuli, including hyperglycemia, elevated free fatty acids, ROS, tumor necrosis factor-α (TNF-α) and other proinflammatory cytokines, p38 MAP kinase and ultraviolet irradiation[82]. NF-κB plays a crucial role in mediating immune and inflammatory responses and apoptosis. Alterations in NF-κB signaling are associated with a number of chronic diseases, such as diabetes and atherosclerosis. The c-jun NH(2)-terminal kinases (JNK) and p38 MAPKs are members of the complex superfamily of MAP serine/threonine protein kinases and are known as stress-activated kinases. This is due to the fact that the activities of these enzymes are stimulated by a variety of exogenous and endogenous stress-inducing stimuli, including oxidative stress, ROS, hyperglycemia and proinflammatory cytokines[85]. JNK is activated by hyperglycemia-induced oxidative stress and is probably involved in apoptosis, which can be suppressed by the antioxidant vitamin C[86] and enhanced by angiotensin II[87]. p38 MAPK is also activated in response to hyperglycemia and in diabetes. In the glomeruli of STZ-induced diabetic rats, p38 MAPK activity was increased compared with control rats, followed by increased phosphorylation of heat shock protein (HSP) 25, a downstream substrate of p38 MAPK. These effects appeared to be the result of increased ROS production[88]. Taken together, these data suggest that NF-κB, JNK and p38 MAPK pathways are potential stress-signaling systems that can lead to late complications of diabetes.
Nitric oxide is an important regulator of cardiac function. However, NO may react with the surrounding O2– to form peroxynitrite (ONOO–). Peroxynitrite, a very active radical similar to the hydroxyl radical, interacts with cytoplasmic proteins to form nitrotyrosine, which has been indicated as a marker for reactive nitrogen species-induced oxidative damage under in vivo conditions[21,89]. It has been suggested that excessive NO is pathophysiological because of its ability to form pro-oxidants. Cardiac NO production and NOS protein levels have been found to be elevated in the hearts of diabetic animals[90]. This is consistent with the observation of a significant increase in nitrotyrosine concentration in myocytes in the hearts of diabetic mice[21].
Role of antioxidants in diabetic cardiomyopathy
Mitochondrial damage is related to ROS formation and plays an important role in the development of diabetic cardiomyopathy[91,92]. Coenzyme Q (CoQ) is an important component in mitochondrial energy metabolism and is also a potent endogenous antioxidant in vivo. In heart mitochondrial preparations of diabetic rats, the concentration of α-tocopherol was increased; however, the concentration of both CoQ-9 and CoQ-10 was decreased[92]. Data from our group have shown that the reduction in coenzyme levels from diabetic animals is attenuated with the supplementation of insulin-like growth factor I (IGF-1)[19].
It is important to note that contractile function of the heart requires a high metabolic demand, and the mitochondrial respiratory chain is the primary energy-releasing system in the myocyte. Through the respiratory chain, a series of oxidation-reduction reactions continually take place in the myocyte. Therefore, an efficient antioxidant system, including superoxide dismutase (SOD), catalase, glutathione peroxidase (GPX), glutathione (GSH) and α-tocopherol are critical to effective functioning of the myocardium. However, in experimental animal models, the heart levels of these antioxidants are much lower than in other organ systems, even in non-diabetic normals[93,94]. In addition, hyperglycemia can impair and decrease the amount of antioxidants within the heart of a diabetic animal[95–97] making it more vulnerable to ROS-induced damage.
The increase in ROS serves to decrease the antioxidant capacity of the diabetic myocardium, contributing significantly to oxidative stress and resultant myocardial damage. This damage causes cardiac morphological and functional abnormalities. Epstein and colleagues[98,99] showed that type 1 diabetic cardiomyopathy could be prevented when the antioxidants metallothionein (MT) and catalase were overexpressed specifically in the heart. They also showed that ROS production was enhanced in genetically diabetic mice (OVE26), which could be prevented by genetically crossing the diabetic mice with those overexpressing the MT or catalase genes[98,99].
Cell death is an important determinant of cardiac remodeling because it causes a loss of contractile units, compensatory hypertrophy of myocardial cells and reparative fibrosis[17]. Apoptotic cell death associated with increased oxidative stress in multiple organ systems of diabetes mellitus has been well documented[100–102]. Recent in vivo experiments have demonstrated the induction of myocardial cell apoptosis in experimental diabetic rats[103], mice[21] and diabetic patients[89]. Heart specimens from diabetic patients (both hypertensive and non-hypertensive) showed an increase in myocyte, endothelial and fibroblast apoptosis[89]. The increased cell death was associated with an increase in ROS formation[21,89,103]. However, the precise mechanism(s) by which ROS accumulation leads to compromised heart function and the effect of antioxidant therapy in diabetic subjects is largely unknown. Therefore, it is important to study the signaling pathways and molecular mechanisms by which hyperglycemia-induced (or, presumably, STZ-induced) oxidative stress leads to cell death and myocardial pathogenesis.
Role of the renin-angiotensin system in the development of diabetic cardiomyopathy
The renin-angiotensin system (RAS) is known to play a major role in the regulation of blood pressure and other functions of the cardiovascular system[104]. Enhanced RAS is implicated in the development of diabetic cardiomyopathy and other heart dysfunctions including coronary insufficiency, congestive heart failure and hypertensive cardiomyopathy[105]. The growth-promoting effects of angiotensin II are mediated primarily through its type 1 receptor (AT1) and the action of RAS is speculated to contribute to diabetic cardiomyopathy[105]. It has been shown that stimulation of the AT1 receptor generates oxygen-derived free radicals, having detrimental effects on the cardiovascular system[106,107]. The AT1 receptor has been shown to be coupled to several postreceptor signaling pathways, including Janus kinase (JAK)/signal transducer and activator of transcription (STAT) and NADPH oxidase[104,108,109]. However, the precise role of RAS, in particular the AT1 receptor, in the development of diabetic cardiomyopathy is still speculative and further study is warranted.
Future directions
Diabetic cardiomyopathy is a clinical problem that is present in both type 1 and type 2 diabetes, which potentially involves myocyte death and interstitial fibrosis. These myocyte and non-myocyte alterations may contribute to compromised ventricular function in diabetes, which is one of the leading causes of death in the world today. It is critical to investigate the underlying (myocyte) causes of diabetic cardiomyopathy and the synergistic impact of oxidative stress in combination with antioxidant therapy on the development of heart dysfunction associated with diabetes.
Acknowledgements
The work in our laboratory has been supported in part by the National Institutes of Health, American Diabetes Association, American Heart Association (Northland and Pacific Mountain Affiliates) and the Max Baer Heart Foundation.
References
- Poulter N. Global risk of cardiovascular disease. Heart 2003;89:2-5.
- Strandberg TE, Salomaa V. Factors related to the development of diabetes during a 20-year follow-up. A prospective study in a homogeneous group of middle-aged men. Nutr Metab Cardiovasc Dis 2000;10:239-46.
- Garcia MJ, McNamara PM, Gordon T, Kannel WB. Morbidity and mortality in diabetics in the Framingham population. Sixteen year follow-up study. Diabetes 1974;23:105-11.
- Sowers JR. Diabetes mellitus and cardiovascular disease in women. Arch Intern Med 1998;158:617-21.
- Pradhan AD, Skerrett PJ, Manson JE. Obesity, diabetes, and coronary risk in women. J Cardiovasc Risk 2002;9:323-30.
- Charpentier G, Genes N, Vaur L, Amar J, Clerson P, Cambou JP, et al. Control of diabetes and cardiovascular risk factors in patients with type 2 diabetes: a nationwide French survey. Diabetes Metab 2003;29:152-8.
- Li S, Culver B, Ren J. Benefit and risk of exercise on myocardial function in diabetes. Pharmacol Res 2003;48:127-32.
- Hammes HP, Du X, Edelstein D, Taguchi T, Matsumura T, Ju Q, et al. Benfotiamine blocks three major pathways of hyperglycemic damage and prevents experimental diabetic retinopathy. Nat Med 2003;93:294-9.
- Feldman EL. Oxidative stress and diabetic neuropathy: a new understanding of an old problem. J Clin Invest 2003;111:431-3.
- Gilbert RE, Tsalamandris C, Bach LA, Panagiotopoulos S, O’Brien RC, Allen TJ, et al. Long-term glycemic control and the rate of progression of early diabetic kidney disease. Kidney Int 1993;44:855-9.
- Fein FS, Sonnenblick EH. Diabetic cardiomyopathy. Cardiovasc Drugs Ther 1994;8:65-73.
- Ren J, Davidoff AJ. Diabetes rapidly induces contractile dysfunctions in isolated ventricular myocytes. Am J Physiol Heart Circ Physiol 1997;272:H148-58.
- Wold LW, Relling DP, Colligan PB, Scott GI, Hintz KK, Ren BH, et al. Characterization of contractile function in diabetic hypertensive cardiomyopathy in adult rat ventricular myocytes. J Mol Cell Cardiol 2001;33:1719-26.
- Poirier P, Garneau C, Marois L, Bogaty P, Dumesnil JG. Diastolic dysfunction in normotensive men with well-controlled type 2 diabetes: importance of maneuvers in echocardiographic screening for preclinical diabetic cardiomyopathy. Diabetes Care 2001;24:5-10.
- Buyukgebiz A, Saylam G, Dundar B, Bober E, Unal N, Akcoral A. Dilated cardiomyopathy as the first early complications in a 14 year-old girl with diabetes mellitus type 1. J Pediatr Endocrinol Metab 2000;13:1143-6.
- Depre C, Young ME, Ying J, Ahuja HS, Han Q, Garza N, et al. Streptozotocin-induced changes in cardiac gene expression in the absence of severe contractile dysfunction. J Mol Cell Cardiol 2000;32:985-96.
- Kang YJ. Molecular and cellular mechanisms of cardiotoxicity. Environ Health Perspect 2001;109:27-34.
- Ren J, Bode AM. Altered cardiac excitation-contraction coupling in ventricular myocytes from spontaneously diabetic BB rats. Am J Physiol Heart Circ Physiol 2000;279:H238-44.
- Norby FL, Wold LE, Duan J, Hintz KK, Ren J. IGF-I attenuates diabetes-induced cardiac contractile dysfunction in ventricular myocytes. Am J Physiol Endocrinol Metab 2002;283:E658-66.
- Norby FL, Aberle NS, Kajstura J, Anversa P, Ren J. Transgenic overexpression of insulin-like growth factor I prevents streptozotocin-induced cardiac contractile dysfunction and beta-adrenergic response in ventricular myocytes. J Endocrinol 2004;180:175-82.
- Kajstura J, Fiordaliso F, Andreoli AM, Li B, Chimenti S, Medow MS, et al. IGF-1 overexpression inhibits the development of diabetic cardiomyopathy and angiotensin II-mediated oxidative stress. Diabetes 2001;50:1414-24.
- Sowers JR, Williams M, Epstein M, Bakris G. Hypertension in patients with diabetes. Strategies for drug therapy to reduce complications. Postgrad Med 2000;107:47-54.
- Spector KS. Diabetic cardiomyopathy. Clin Cardiol 1998;21:885-7.
- Rubler S, Dlugash J, Yuceoglu YZ, Kumral T, Branwood AW, Grishman A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am J Cardiol 1972;30:595-602.
- Pierce GN, Russell JC. Regulation of intracellular Ca2+ in the heart during diabetes. Cardiovasc Res 1997;34:41-7.
- Chatham JC, Forder JR, McNeill JH. The heart in diabetes. Norwell: MA Kluwer Academic Publishers; 1996.
- Schaffer SW, Mozafferi M. Abnormal mechanical function in diabetes: relation to myocardial calcium handling. Coron Artery Dis 1996;7:109-15.
- Dincer UD, Bidasee KR, Guner S, Tay A, Ozcelikay AT, Altan VM. The effect of diabetes on expression of beta1-, beta2-, and beta3-adrenoreceptors in rat hearts. Diabetes 2001;50:455-61.
- Tamada A, Hattori Y, Houzen H, Yamada Y, Sakuma I, Kitabatake A, et al. Effects of beta-adrenoreceptor stimulation on contractility, [Ca2+]i, and Ca2+ current in diabetic rat cardiomyocytes. Am J Physiol Heart Circ Physiol 1998;274:H1849-57.
- Idris I, Gray S, Donnelly R. Protein kinase C activation: isozyme-specific effects on metabolism and cardiovascular complications in diabetes. Diabetologia 2001;44:659-73.
- Given MB, Jie O, Zhao X, Giles TD, Greenberg SS. Protein kinase C isozymes in skeletal muscles during the early stage of genetic and streptozotocin diabetes. Proc Soc Exp Biol Med 1998;218:382-9.
- Devereux RB, Roman MJ, Paranicas M, O’Grady MJ, Lee ET, Welty TK, et al. Impact of diabetes on cardiac structure and function: the strong heart study. Circulation 2000;101:2271-6.
- Liu JE, Palmieri V, Roman MJ, Bella JN, Fabsitz R, Howard BV, et al. The impact of diabetes on left ventricular filling pattern in normotensive and hypertensive adults: the Strong Heart Study. J Am Coll Cardiol 2001;37:1943-9.
- Rutter MK, Parise H, Benjamin EJ, Levy D, Larson MG, Meigs JB, et al. Impact of glucose intolerance and insulin resistance on cardiac structure and function: sex-related differences in the Framingham Heart Study. Circulation 2003;107:448-54.
- Schannwell CM, Schneppenheim M, Perings SM, Zimmermann T, Plehn G, Strauer BE. Alterations of left ventricular function in women with insulin-dependent diabetes mellitus during pregnancy. Diabetologia 2003;46:267-75.
- Dutta K, Podolin DA, Davidson MB, Davidoff AJ. Cardiomyocyte dysfunction in sucrose-fed rats is associated with insulin resistance. Diabetes 2001;50:1186-92.
- Zarich SW, Nesto RW. Diabetic cardiomyopathy. Curriculum Cardiol 1989;118:1000-12.
- Fraser GE, Luk R, Thompson S, Smith H, Carter S, Sharpe N. Comparison of echocardiography variables between Type 1 diabetics and normal control. Am J Cardiol 1995;75:141-5.
- Raev DC. Which left ventricular function is impaired earlier in the evaluation of diabetic cardiomyopathy. Diabetes Care 1994;17:633-9.
- Celentano A, Vaccaro O, Tammaro P, Galderisi M, Crivaro M, Oliviero M, et al. Early abnormalities of cardiac function in non-insulin-dependent diabetes mellitus and impaired glucose tolerance. Am J Cardiol 1995;76:1173-6.
- Grundy SM, Benjamin IJ, Burke GL, Chait A, Eckel RH, Howard BV, et al. Diabetes and cardiovascular disease: a statement for healthcare professionals from the American Heart Association. Circulation 1999;100:1134-46.
- Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001;414:813-20.
- Cai L, Kang YJ. Oxidative stress and diabetic cardiomyopathy: a brief review. Cardiovasc Toxicol 2001;1:181-93.
- Ramasamy R, Oates PJ, Schaefer S. Aldose reductase inhibition protects diabetic and nondiabetic rat hearts from ischemic injury. Diabetes 1997;46:292-300.
- Hwang YC, Bakr S, Ellery CA, Oates PJ, Ramasamy R. Sorbitol dehydrogenase: a novel target for adjunctive protection of ischemic myocardium. FASEB J 2003;17:2331-3.
- Ramasamy R. Aldose reductase: a novel target for cardioprotective interventions. Curr Drug Targets 2003;4:625-32.
- Chandra D, Jackson EB, Ramana KV, Kelley R, Srivastava SK, Bhatnagar A. Nitric oxide prevents aldose reductase activation and sorbitol accumulation during diabetes. Diabetes 2002;51:3095-101.
- Lee AY, Chung SS. Contributions of polyol pathway to oxidative stress in diabetic cataract. FASEB J 1999;13:23-30.
- Makita Z, Vlassara H, Rayfield E, Cartwright K, Friedman E, Rodby R, et al. Hemoglobin-AGE: a circulating marker of advanced glycosylation. Science 1992;258:651-3.
- Wolffenbuttel BH, Giordano D, Founds HW, Bucala R. Long-term assessment of glucose control by haemoglobin-AGE measurement. Lancet 1996;347:513-5.
- Bierhaus A, Illmer T, Kasper M, Luther T, Quehenberger P, Tritschler H, et al. Advanced glycation end product (AGE)-mediated induction of tissue factor in cultured endothelial cells is dependent on RAGE. Circulation 1997;96:2262-71.
- Bierhaus A, Hofmann MA, Ziegler R, Nawroth PP. AGEs and their interation with AGE-receptors in vascular disease and diabetes mellitus. I. The AGE concept. Cardiovasc Res 1998;37:586-600.
- Schmidt AM, Stern DM. RAGE: a new target for the prevention and treatment of the vascular and inflammatory complications of diabetes. Trends Endocrinol Metab 2000;11:368-75.
- Esposito C, Gerlach H, Brett J, Stern D, Vlassara H. Endothelial receptor-mediated binding of glucose-modified albumin is associated with increased monolayer permeability and modulation of cell surface coagulant properties. J Exp Med 1989;170:1387-407.
- Li J, Schmidt AM. Characterization and functional analysis of the promoter of RAGE, the receptor for advanced glycation end products. J Biol Chem 1997; 272: 16 498–506.
- Schmidt AM, Hori O, Brett J, Yan SD, Wautier JL, Stern D. Cellular receptors for advanced glycation end products. Implications for induction of oxidant stress and cellular dysfunction in the pathogenesis of vascular lesions. Arterioscler Thromb 1994;14:1521-8.
- Ritthaler U, Deng Y, Zhang Y, Greten J, Abel M, Sido B, et al. Expression of receptors for advanced glycation end products in peripheral occlusive vascular disease. Am J Pathol 1995;146:688-94.
- Yan SD, Stern D, Schmidt AM. What’s the RAGE? The receptor for advanced glycation end products (RAGE) and the dark side of glucose. Eur J Clin Invest 1997;27:179-81.
- Lu M, Kuroki M, Amano S, Tolentino M, Keough K, Kim I, et al. Advanced glycation end products increase retinal vascular endothelial growth factor expression. J Clin Invest 1998;101:1219-24.
- Soudis-Liparota T, Cooper M, Papazoglou D, Clarke B, Jerums G. Retardation by aminoguanidine of development of albuminura, mesangial expansion, and tissue fluorescence in streptozotocin-induced diabetic rat. Diabetes 1991;40:1328-34.
- Nakamura S. Progression of nephropathy in spontaneous diabetic rats is prevented by OPB-9195, a novel inhibitor of advanced glycation. Diabetes 1997;46:895-9.
- Hammes HP. Aminoguanidine treatment inhibits the development of experimental diabetic retinopathy. Proc Natl Acad Sci USA 1991;88:11555-9.
- Park L. Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nature Med 1998;4:1025-31.
- Koya D, King GL. Protein kinase C activation and the development of diabetic complications. Diabetes 1998;47:859-66.
- Portilla D. Etomoxir-induced PPARalpha-modulated enzymes protect during acute renal failure. Am J Physiol Renal Physiol 2000;278:F667-75.
- Keogh RJ, Dunlop ME, Larkins RG. Effect of inhibition of aldose reductase on glucose flux, diacylglycerol formation, protein kinase C, and phospholipase A2 activation. Metabolism 1997;46:41-7.
- Stevens MJ, Obrosova I, Feldman EL, Greene DA. The sorbitol-osmotic and sorbitol-redox hypothesis. In: LeRoith D, Taylor SI, Olefsky JM, editors. Diabetes mellitus: a fundamental and clinical text. Philadelphia: Lippincott Williams & Wilkins; 2000. p 972–83.
- Lee AY, Chung SK, Chung SS. Demonstration that polyol accumulation is responsible for diabetic cataract by the use of transgenic mice expressing the aldose reductase gene in the lens. Proc Natl Acad Sci USA 1995;92:2780-4.
- Yamaoka T, Nishimura C, Yamashita K, Itakura M, Yamada T, Fujimoto J, et al. Acute onset of diabetic pathological changes in transgenic mice with human aldose reductase cDNA. Diabetologia 1995;38:255-61.
- Yagihashi S, Yamagishi S, Wada R, Sugimoto K, Baba M, Wong HG, et al. Galactosemic neuropathy in transgenic mice for human aldose reductase. Diabetes 1996;45:56-9.
- Singh SB, Malamas MS, Hohman TC, Nilakantan R, Carper DA, Kitchen D. Molecular modeling of the aldose reductase-inhibitor complex based on the X-ray crystal structure and studies with single-site-directed mutants. J Med Chem 2000;43:1062-70.
- Du XL. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proc Natl Acad Sci USA 2000;97:12222-6.
- Kakkar R, Kalra J, Mantha SV, Prasad K. Lipid peroxidation and activity of antioxidant enzymes in diabetic rats. Mol Cell Biochem 1995;151:113-9.
- Ohuwa T, Sato Y, Naoi M. Hydroxyl radical formation in diabetic rats induced by streptozotocin. Life Sci 1995;56:1789-98.
- Bayes JW, Thorpe SR. Role of oxidative stress in diabetic complications: a new perspective on an old paradigm. Diabetes 1999;48:1-9.
- Kowluru RA, Engerman RL, Kern TS. Diabetes-induced metabolic abnormalities in myocardium: effect of antioxidant therapy. Free Radical Res 2000;32:67-74.
- Ustinova EE, Barrett CJ, Sun SY, Schultz HD. Oxidative stress impairs cardiac chemoreflexes in diabetic rats. Am J Physiol 2000;279:2176-87.
- Rosen P, Du X, Sui GZ. Molecular mechanisms of endothelial dysfunction in the diabetic heart. Adv Exp Med Biol 2001;498:75-86.
- Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000;404:787-90.
- Mohamed AK, Bierhaus A, Schiekofer S, Tritschler H, Ziegler R, Nawroth PP. The role of oxidative stress and NF-κB activation in late diabetic complications. Biofactors 1999;10:157-67.
- Mercurio F, Manning AM. NF-κB as a primary regulator of the stress response. Oncogene 1999;18:6163-71.
- Barnes PJ, Karin M. Nuclear factor-κB: a pivotal transcription factor in chronic inflammatory disease. N Engl J Med 1997;336:1066-71.
- Baldwin AS Jr. The transcription factor NF-κB and human disease. J Clin Invest 2001;107:3-6.
- Tak PP, Firestein GS. NF-κB: a key role in inflammatory disease. J Clin Invest 2001;107:7-11.
- Tibbles LA, Woodgett JR. The stress-activated protein kinase pathways. Cell Mol Life Sci 1999;55:1230-54.
- Ho FM, Liu SH, Liau CS, Huang PJ, Lin-Shiau SY. High glucose-induced apoptosis in human endothelial cells is mediated by sequential activations of c-Jun NH(2)-terminal kinase and caspase-3. Circulation 2000;101:2618-24.
- Natarajan R, Scott S, Bai W, Yerneni KKV, Nadler J. Angiotensin II signaling in vascular smooth muscle cells under high glucose conditions. Hypertension 1999;33:378-84.
- Dunlop ME, Muggli EE. Small heat shock protein alteration provides a mechanism to reduce mesangial cell contractility in diabetes and oxidative stress. Kidney Intl 2000;57:464-75.
- Frustaci A, Kajstura J, Chimenti C, Jakoniuk I, Leri A, Maseri A, et al. Myocardial cell death in human diabetes. Circ Res 2000;87:1123-32.
- Stockklauser-Farber K, Ballhausen T, Laufer A, Rosen P. Influence of diabetes on cardiac nitric oxide synthase expression and activity. Biochim Biophys Acta 2000;1535:10-20.
- Tomita M, Mukae S, Geshi E, Umetsu K, Nakatani M, Katagiri T. Mitochondrial respiratory impairment in streptozotocin-induced diabetic rat heart. Jpn Circ J 1996;60:673-82.
- Kucharska J, Braunova Z, Ulicna O, Zlatos L, Gvozdjakova A. Deficit of coenzyme Q in heart and liver mitochondria of rats with streptozotocin-induced diabetes. Physiol Res 2000;49:411-8.
- Doroshow JH, Locker GY, Myers CE. Enzymatic defenses of the mouse heart against reactive oxygen metabolites: alterations produced by doxorubicin. J Clin Invest 1980;65:128-35.
- Chen Y, Saari JT, Kang YJ. Weak antioxidant defenses make the heart a target for damage in copper-deficient rats. Free Radical Biol Med 1994;17:529-36.
- Kersten JR, Schmeling TJ, Orth KG, Pagel PS, Warltier DC. Acute hyperglycemia abolishes ischemic preconditioning in vivo. Am J Physiol 1998;275:H721-5.
- Joyeux M, Faure P, Godin-Ribuot D, Halimi S, Patel A, Yellon DM, et al. Heat stress fails to protect myocardium of streptozotocin-induced diabetic rats against infarction. Cardiovasc Res 1999;43:939-46.
- Elangovan V, Shohami E, Gati I, Kohen R. Increased hepatic lipid soluble antioxidant capacity as compared to other organs of streptozotocin-induced diabetic rats: a cyclic voltametry study. Free Radical Res 2000;32:125-34.
- Ye G, Metreveli NS, Ren J, Epstein PN. Metallothionein prevents diabetes-induced deficits in cardiomyocytes by inhibiting reactive oxygen species production. Diabetes 2003;52:777-83.
- Ye G, Metreveli NS, Donthi RV, Xia S, Xu M, Carlson EC, et al. Catalase protects cardiomyocyte function in models of type 1 and type 2 diabetes. Diabetes 2004;53:1336-43.
- Alici B, Gumustas MK, Ozkara H, Akkus E, Demirel G, Yencilek F, et al. Apoptosis in the erectile tissues of diabetic and healthy rats. BJU Int 2000;85:326-9.
- Cai L, Chen S, Evans T, Deng DX, Mukherjee K, Chakrabarti S. Apoptotic germ-cell death and testicular damage in experimental diabetes: prevention by endothelin antagonism. Urol Res 2000;28:342-7.
- Srinivasan S, Stevens M, Wiley JW. Diabetic peripheral neuropathy: evidence for apoptosis and associated mitochondrial dysfunction. Diabetes 2000;49:1932-8.
- Fiordaliso F, Li B, Latini R, Sonnenblick EH, Anversa P, Leri A, et al. Myocyte death in streptozotocin-induced diabetes in rats is angiotensin II-dependent. Lab Invest 2000;80:531-7.
- Privratsky JR, Wold LE, Sowers JR, Quinn MR, Ren J. AT1 blockade prevents glucose-induced cardiac dysfunction in ventricular myocytes: role of the AT1 receptor and NADPH oxidase. Hypertension 2003;42:206-12.
- Dzau VJ. Theodore Cooper Lecture: tissue angiotensin and pathobiology of vascular disease: a unifying hypothesis. Hypertension 2001;37:1047-52.
- Nickenig G, Harrison DG. The AT1-type angiotensin receptor in oxidative stress and atherogenesis, part I: oxidative stress and atherogenesis. Circulation 2002;105:393-6.
- Sowers JR. Hypertension, angiotensin II, and oxidative stress. N Engl J Med 2002;346:1999-2001.
- Mascareno E, Siddiqui MA. The role of Jak/STAT signaling in heart tissue renin-angiotensin system. Mol Cell Biochem 2000;212:171-5.
- Wang HD, Xu S, Johns DG, Du Y, Quinn MT, Cayatte AJ, et al. Role of NADPH oxidase in the vascular hypertrophic and oxidative stress response to angiotensin II in mice. Circ Res 2001;88:947-53.