
Protective effect of membrane cofactor protein against complement-dependent injury
Introduction
The first barrier to a successful xenograft is the hyperacute rejection (HAR), that is initiated by the activation of complement on the graft’ vessel walls. The fragments of activated complement, such as C3a, C5a and membrane attack complex (MAC), promote blood coagulation and induce endothelial cell injury. Although drugs with anti-complement activity, such as heparin and FUT-175, have been used to prevent thrombosis, their clinical application is limited because they interfere with host blood coagulation or have other toxic effects. If the activation of complement is blocked, intravascular coagulation can be prevented effectively.
Membrane cofactor protein (MCP, CD46) belongs to the complement regulator protein family. It is located on the cellular membrane of all nucleated cells. MCP inhibits the activation of complement by combining with C3b/C4b and serves as a cofactor in their proteolytic cleavage by complement serine protease factor I. Generally, MCP function downregulates complement activation. Therefore, it is possible to prevent lesions caused by abnormal activated complement[1].
In this study, cobra venom factor (CVF), zymosan A and the ovalbumin (OVA)–anti-OVA complex were used as complement activators to observe the protective effect of MCP on complement-dependent lesions.
Materials and methods
Reagents and animals Cobra venom factor (CVF) was isolated and purified by successive column chromatography from the venom of Naja naja atra, as described previously[2]. DEAE-sephadex A-50 was purchased from Pharmacia, USA; β-mercaptoethanol, Coomassie brilliant blue R-250 and Nonidet P-40 were from Fluka, USA; acrylamide and N,N-methylene bisacrylamide was from Bio-Rad, USA; sodium azide, Evans blue dye, zymosan A and OVA were from Sigma, USA; rabbit anti-OVA from Chemicon; and benzanidine and phenylmethyl sulfonyl fluoride (PMSF) were from Amresco, USA. All chemicals were of analytical grade.
Blood was freshly obtained from pigs or sheep. O-type human plasma was purchased from the blood bank of the First Affiliated Hospital of Sun Yat-Sen University. Sprague-Dawley rats (Grade II, Certificate N
Isolation of membrane proteins from pig erythrocyte ghosts Pig erythrocytes were washed 3 times with phosphate buffered solution (PBS, in mmol: Na2HPO4 10.1, KH2PO4 1.7, NaCl 137, KCl 2.7; pH 7.4) and resuspended (1/10) in lysis buffer [in mmol: Na2HPO4 5, edetic acid (EDTA) 2, benzanidine 1, Phenylmethylsulfonyl Fluoride (PMSF) 1, sodium azide 0.05%; pH 7.4]. After being stirred overnight at 4 oC, the erythrocyte ghosts were concentrated (12 000×g, 4 oC, 45 min), and washed again with lysis buffer (1:1) and centrifuged (12 000×g, 4 oC, 45 min). The erythrocyte ghosts were then diluted 1/5 in PBS and extracted with 0.1% Nonidet P-40 at 4 oC for 1 h. After centrifugation (12 000×g, 4 oC, 30 min), the erythrocyte ghost extract was added to the top of a DEAE sephadex A-50 column (∅: 2.6 cm; L: 25 cm) and washed with PBS containing a NaCl concentration gradient from 0 to 1 mol/L. The flow-rate was 24 mL/h. The fraction corresponding to the second ultraviolet absorbance peak, which possessed anti-complement activity, was collected and stored.
Identification of MCP The molecular weight of MCP was determined according to Rf on reduced SDS-PAGE, stained with Coomassie brilliant blue R-250. CH50 was determined according to the absorbance of sensitized sheep erythrocytes incubated with fresh guinea pig plasma, to which a series of concentrations of MCP were added. Protein concentrations were determined by using the Coomassie brilliant blue R-250 method.
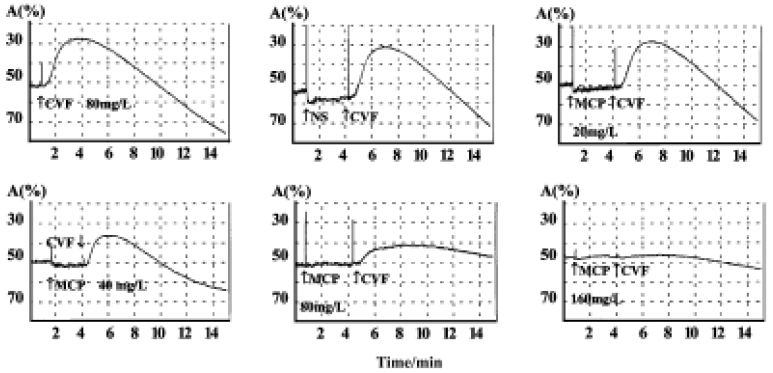
Platelet metamorphosis and aggregation The metamorphosis and aggregation of male guinea pig platelets in platelet rich plasma (PRP) were recorded by an aggrometer (Chrono-Log, USA) linked to a computer with an A/D converter. Platelet poor plasma (PPP) was used for a control. After the baseline was steady, MCP (0, 20, 40, 80, and 160 mg/L) was added to PRP 3 min before CVF (80 mg/L) to observe the aggregation line changes.
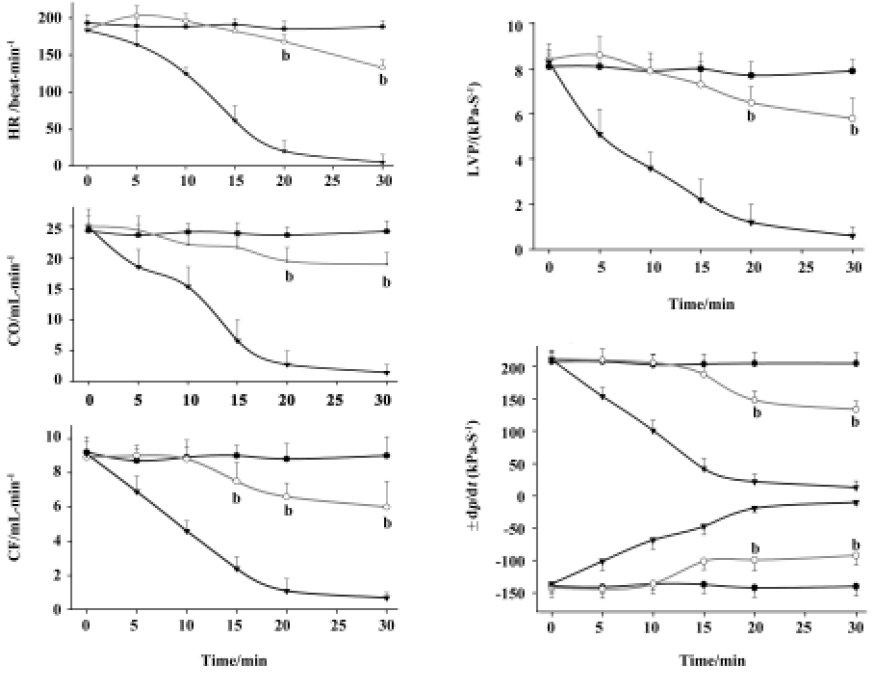
Isolated working guinea pig heart Male guinea pigs weighing 380 g±35 g (n=15) were divided randomly into three groups. The guinea pig hearts were removed quickly and then perfused in a retrograde manner with modified Krebs-Henseleit buffer (in mmol: NaCl 11.2, KCl 5.7, MgCl2 1.2, CaCl2 2.5, NaHCO3 26.2, Na2-EDTA 0.5, glucose 11.0; pH 7.2) at 37 oC, saturated by a mixture of 95% O2 and 5% CO2. After the heart began to beat again, a plastic cannula was inserted into the left atrium, and retrograde perfusion was changed to anterograde. Left ventricular pressure (LVP) was detected by a pressure transducer connected to the canal, which was inserted into the left ventricular cave. LVP, dp/dt, and heart rate (HR) were recorded by a polygraph (LMS-2A, Shanghai, China). Aortic flow (AF) was measured by an electromagnetic flowmeter (MFV-1200, Nihon Kohden, Japan), and coronary flow (CF) was measured manually. Cardiac output (CO) was defined as the sum of AF and CF.
After the CF became steady at 8 to 9 mL per min, we carried out the following treatment: 56 oC preheated human plasma was added into perfusate in the negative control group. Human plasma pre-incubated with zymosan A was used for the positive control group. In group 3, human plasma was incubated with both MCP and zymosan A before being added.
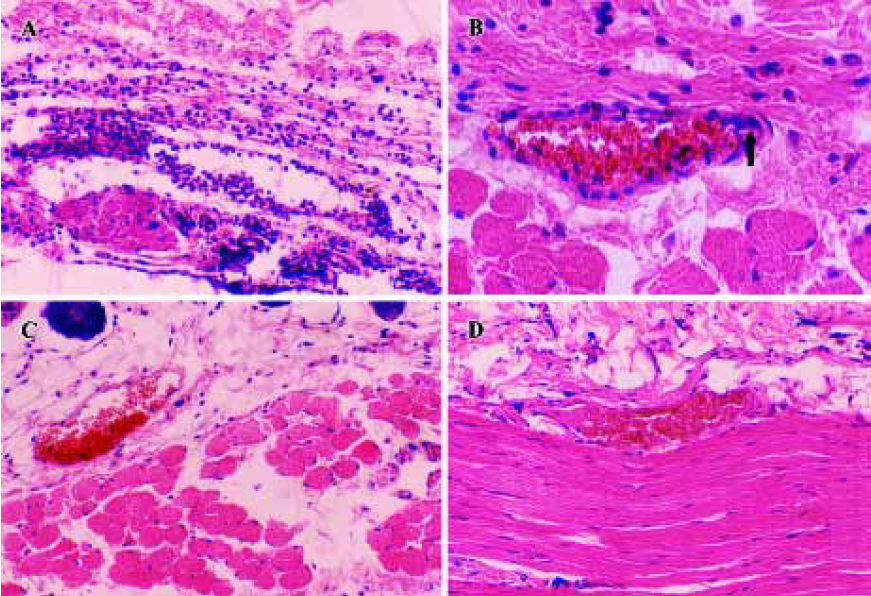
Reverse passive Arthus reaction Anesthetized female rats (140 g–170 g, n=10) were given 0.6 mL of 0.9% NaCl solution iv, which contained 5 mg OVA and 2% Evans blue dye. Five minutes later, 0.1 mL PBS containing 0.5 mg rabbit-anti-OVA IgG with or without 0.4 mg MCP was injected sc on both sides of the back. Six hours later, rats were killed, and the area of dye leakage was measured. Skin samples were dissected, fixed with formalin, and then stained with hematoxylin and eosin.
Statistical analysis Statistical tests were performed using the t-test of SigmaPlot software. P<0.05 was considered to be statistically significant.
Results
Isolation of MCP A total of 2.1 mg MCP was gained from 100 mL pig erythrocytes. The molecular weight of MCP was determined to be 51 kDa according to the Rf value from SDS-PAGE. One anti-complement activity unit (CH50) of MCP was 39.8±0.7 µg
Inhibition of CVF-induced platelet metamorphosis and aggregation MCP, at concentrations from 20 mg/L to 160 mg/L, dose-dependently inhibited the rat platelet metamorphosis induced by CVF (Figure 1). The linear regression equation was Y=89.3lgX–106.6 (where Y is the inhibitory ratio percentage, and X is the concentration of MCP; r=0.98, n=20, P<0.05). The IC50 was 56.7±2.6 mg/L. MCP had little effect on CVF-induced platelet aggregation.
Protection of human complement-induced lesions on isolated working hearts Cardiac function parameters, namely LVP, dp/dt, CO, CF, AF, and HR, remained relatively stable for about 2 h in 56 oC inactivated complement treatment (negative control group). In positive control group, hearts quickly became non-functional after zymosan A (16.7 mg/L) activated complement was added. CF, CO, LVP and dp/dt decreased markedly after 5 min (Figure 2; P<0.05 vs negative control group, n=5), and declined to their lowest levels at 30 min. Yet when 1.3 mg/L MCP was co-incubated with zymosan A and complement, no change was seen in the first 20 min (P<0.05 vs negative control group, n=5).
Reverse passive Arthus reaction In our study, vigorous infiltration of polymorphonuclear cells, perivascular cuffing and mini clots were seen in the skin lesions under light microscopy (Figure 3A,3B), and the skin lesion area (dye leakage indicating edema) was 330 mm2±15 mm2 in size (n=5). The skin lesion area in the MCP-treated group, 149 mm2±10 mm2 in size, was smaller (P<0.05 vs control, n=5). The inflammatory reaction (cellular infiltrate) was less than that in the control group and no clotting or perivascular cuffing was found (Figure 3C, 3D), which suggests that MCP significantly reduced the extent of inflammatory injury.
Discussion
MCP is widely distributed on epithelial and endothelial cells, cells originating from fibroblasts, and all animal blood cells except human erythrocytes[3]. Because it is more abundantly expressed on erythrocytes from pigs than on those from other animals, the MCP used in this study was derived from pig erythrocytes. Structurally, pig-derived MCP amino acid sequence has 43% homology with human-derived MCP[4], and it has no N-glycosylation site in the second repeated motif (short consensus repeat, SCR), which serves as a measles receptor. Because it is a safer complement inhibiting factor than human-derived MCP, pig-derived MCP was worth testing for its protective action in complement-dependent injury. The DEAE sephadex A-50 anion exchange column is able to separate proteins by both their charge and molecular weight. Although one-step anion exchange separation can result in only impure proteins, in our study, the pig-derived MCP did shown the complement regulation ability, more importantly, the high yield made continuation of our experiment possible.
CVF can activate complement through an alternative pathway: its terminal product MAC can induce platelet metamorphosis and express binding sites for the coagulation factors Va and Xa on platelet membranes, triggering coagulation[5]. In the present study, MCP inhibited the platelet metamorphosis induced by MAC. Thus, MCP can be used to prevent the intravascular coagulation initiated by activated complement. Decay-accelerating factor (DAF, CD59), another complement-regulating protein, has been reported to have a similar effect[2].
We further investigated the anti-complement effect of MCP in vivo and in vitro. The isolated working heart was an in vitro model of HAR. When zymosan A-treated serum reached the coronary vessel wall, the complement was activated and then cascade responses initiated the HAR, causing the hearts to fail quickly. However, when MCP was pre-incubated, it inhibited the complement activation by zymosan A. Thus, hemodynamic indexes such as LVP, dp/dt, HR, CO and CF, were well maintained. This observation shows that MCP protects the isolated working heart from complement injury. DAF has also been found to have a similar protective effect[6]. The above facts show that downregulating complement activation is an effective way to alleviate the HAR.
The reverse passive Arthus reaction is an in vivo model of HAR, and it refers a type III immune complex mediated hypersensitivity reaction that occurs following the intradermal injection of antigen in the presence of a high level of circulating antibody. In that process, antigen-antibody complex also activated complement. The histological lesions in the MCP-treated group were smaller than those in the control group, suggesting that MCP blocks the classical pathway of complement activation. This finding corresponds with that of an earlier report about blockades preventing monocyte activation and anaphylatoxin damage[7].
Because of its antigenicity, MCP is not completely safe for clinical uses. To improve its safety, further research and chemical modifications are required. Recently, a human transgenic pig model has been established, and it has been shown that HAR in xenotransplantation is suppressed to a large degree[8,9].
In summary, we showed that MCP alleviated complement-mediated injury. Preventing the activation of complement is an effective method to ameliorate such complement-mediated injury.
References
- Song WC. Membrane complement regulatory proteins in autoimmune and inflammatory tissue injury. Curr Dir Autoimmun 2004;7:181-99.
- Gu YJ, Huang SJ, Sun JJ. Fractionation of CD59 from human erythrocyte membrane and protection on complement-dependent platelet injury. Chin Pharma Bull 1998;14:512-15.
- Liszewski MK, Post TW, Atkinson JP. Membrane cofactor protein (MCP or CD46): newest member of the regulators of complement activation gene cluster. Annu Rev Immunol 1991;9:431-55.
- Toyomura K, Fujimura T, Murakami H, Natsume T, Shigehisa T, Inoue N, et al. Molecular cloning of a pig homologue of membrane cofactor protein (CD46). Int Immunol 1997;9:869-76.
- Gorbet MB, Sefton MV. Biomaterial-associated thrombosis: roles of coagulation factors, complement, platelets and leukocytes. Biomaterials 2004;25:5681-703.
- Wu SH, Ma H, Huang SJ, Dong YG, Wu CK, Sun JJ. CD59 prevents human complement-mediated injuries in isolated guinea pig hearts. Chin Med J (Engl) 2002;115:175-8.
- Rinder CS, Rinder HM, Johnson K, Smith M, Lee DL, Tracey J, et al. Role of C3 cleavage in monocyte activation during extracorporeal circulation. Circulation 1999;100:553-8.
- Diamond LE, Quinn CM, Martin MJ, Lawson J, Platt JL, Logan JS. A human CD46 transgenic pig model system for the study of discordant xenotransplantation. Transplantation 2001;71:132-42.
- Loveland BE, Milland J, Kyriakou P, Thorley BR, Christiansen D, Lanteri MB, et al. Characterization of a CD46 transgenic pig and protection of transgenic kidneys against hyperacute rejection in non-immunosuppressed baboons. Xenotransplantation 2004;11:171-83.