
Interleukin-1 beta induction of neuron apoptosis depends on p38 mitogen-activated protein kinase activity after spinal cord injury1
Introduction
Traumatic spinal cord injury (SCI) often results in complete loss of voluntary motor and sensory function below the site of injury. Extensive evidence indicate that secondary injury of the cord involving widespread apoptosis of neurons and oligodendroglia after the trauma contributes significantly to long-term neurological deficits[1,2]. Interleukin-1 beta (IL-1β) induces apoptosis in neurons in vitro[3] and in cultured human astrocytes[4] and oligodendrocytes in vivo[5]. IL-1β expression is upregulated rapidly at the lesion site after SCI[6–8]. Furthermore, injection of an IL-1 receptor antagonist (IL-1Ra), a potent anti-inflammatory cytokine, reduces IL-1β production in the spinal cord and thereby may promote functional recovery following SCI[9]. Thus a rapid accumulation of IL-1β following SCI may initiate apoptosis in neurons and glial cells. The intracellular signaling events initiated by IL-1β that lead to apoptotic cell death in vivo however are not well understood.
Caspase-3 is a potential mediator of apoptosis after central nervous system (CNS) injury[10,11] and its activation may be as a marker of apoptotic cell death. Several studies have provided evidence that cell death from moderate traumatic spinal cord injury is regulated, in part, by apoptosis that involves the caspase family of cysteine proteases[9,11]. In the hippocampus of aged rats, the concentration of IL-1β is increased and this increase is accompanied by enhanced caspase-3 activity indicative of cell death[12]. These findings suggest that neuron apoptosis in the CNS is induced by increased IL-1β through activity of the caspase-3 apoptotic pathway.
p38 Mitogen-activated protein kinase (p38 MAPK) is a pivotal molecule in the signal pathway initiated by IL-1RI[13]. A subset of cyclin-dependent kinase/mitogen-activated protein kinase superfamily kinases has been implicated in neuron apoptosis[14,15]. In neurons of the hippocampus, inhibition of p38 MAPK activation induced by SB203580 blocks suppression of long term potentiation evoked by IL-1β[16] and cellular stress responses to IL-1β[17]. In addition, the cardiovascular and behavioral responses induced by intracerebral ventricular (icv) injection of IL-1β were abrogated by pretreatment with the p38 inhibitor SB203580 (icv) in awake rats[18]. These findings suggest that the effects of IL-1β are partially dependent upon p38 MAPK activation. In addition, IL-1β concentration was found to be increased and accompanied by enhanced caspase-3 activity in the hippocampus of aged rats[12]. Analysis of colocalization of activated caspase-3 with activated p38 MAPK (p-p38) in the hippo-campus of aged rats, suggested that p-p38 was necessary for activation of caspase-3; while in vitro analysis indicated that the IL-1β-induced increase in caspase-3 activity was abrogated by the p38 MAPK inhibitor, SB203580[12]. Thus apoptosis activated by increased IL-1β after SCI may be dependent, at least in part, upon the activation of p38 MAPK.
In the present report, expression of IL-1β as well as expression of its potential downstream target molecules p38 MAPK and caspase-3 was examined in rats following SCI. If p38 MAPK signaling mediates IL-1β genetic induction of apoptosis, then p38 MAPK expression should be elevated after SCI together with IL-1β expression. Likewise, if the apoptotic program initiated by IL-1β involves caspase-3 as an effector of apoptosis, then a subsequent increase in caspase-3 levels should also be observed following SCI.
We further tested pharmacologically whether the apoptotic cascade initiated by IL-1β after SCI involved p38 MAPK signaling and caspase-3. If post-injury caspase-3 increases were induced by IL-1β signaling, then blocking IL-1 receptors should attenuate caspase-3 upregulation after injury. Moreover, if IL-1β induction of apoptosis after SCI was mediated by MAPK signaling, which then resulted in caspase-3 upregulation, then blocking MAPK activity should also attenuate caspase-3 upregulation as well as reduce the amount of apoptotic cells observed after SCI.
Materials and methods
Materials Rabbit antibodies directed against IL-1β, caspase-3 and normal rabbit serum were purchased from Sigma Biotechnologies (Sigma-Aldrich, Inc, Saint Louis, Missouri, USA). p38 MAP Kinase Assay Kits were purchased from Cell Signaling TechnologyTM (Cell Signaling, Beverly, USA). The p38 inhibitor 4-(4-fluorophenyl)-2-(4-methylsulfonylphenyl)-5-(4-pyridyl)-1H-imidazole (SB203580) was purchased from Calbiochem (Calbiochem, La Jolla, CA). Recombinant rat IL-1ra was purchased from R & D Systems (Minneapolis, MN, USA). Terminal Deoxynucleotidyl Transferase-Mediated Deoxyuridine Triphosphate-Biotin Nick End Labeling (TUNEL) detection kits were purchased from Sino-American Biotechnology Co (Sino-American Biotechnology Co, Luo Yang, China).
Spinal cord injury and pharmacological treatment Adult male Wistar rats, weighing about 250 g each (range from 220 g to 270 g), were used in this study (provided by the Experimental Animal Center of Shantou University Medical College). Rats were assigned randomly to either the SCI group, the sham-operated control group (n=54 for each group) or the drug-treated group (including SB203580; IL-1Ra; vehicle, n=18 for each group). Rats’ spinal cords were contused extradurally with a 10-g weight-drop impactor dropped 25.0 mm onto the T9-T10 region of the cord exposed by laminectomy as described previously[19]. Drug treatments were given intrathecally immediately after the injury. Recombinant rat IL-1Ra (10 µg) was given to antagonize IL-1 receptors and SB203580 (4 kg in 10 µL) was given to inhibit p38 MAPK[9,20,21]. The animals in the sham-operated control group underwent a T9 laminectomy without injury and received intrathecal injections of only vehicle (10 µL saline). All animal care and surgical procedures were approved by the institutional animal care and use committee of Shantou University.
RNA purification and RT-PCR At several time points following impact, 4-mm thick tissue sections were collected by deep anesthesia and stored frozen at -80 °C. Total tissue RNA was purified by homogenization of 20 mg spinal cord using the RNeasy Mini kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. RNA was washed, dried, resuspended in sterile nuclease-free water, and stored at -80 °C. After spectrophotometric quantification, the purified RNA was separated on a formaldehyde-agarose gel to assess the extent of degradation.
One microgram total RNA was reverse-transcribed into cDNA in each 50 µL reaction mixture, using one-step RT-PCR (QIAGEN One Step RT-PCR Kit, Qiagen, Valencia, CA) according to the manufacturer’s instructions. The sequences of forward and reverse primers for amplifying IL-1β cDNA were 5'-AGCTATGGCAACTGTCCCT-3' and 5'-AGCTA-TGGCAACTGTCCCT-3', respectively. After amplification, RT-PCR products were quantified with real time PCR.
Real time quantitative RT-PCR To compare the mRNA content of IL-1β in spinal cord after SCI with pharmacological treatment, reverse-transcribed cDNA standards and samples were amplified in parallel by PCR on a LightCycler using additional sequence-specific hybridization probes in combination with the LightCycler. PCR with Taq polymerase (Taqman; Applied Biosystems, Foster City, CA) on the cDNA samples was performed on a sequence detector (Prism 7700; Applied Biosystems, Foster City, CA). Equal amounts of cDNA were used in duplicate and amplified with the PCR mix (Taqman Master Mix; Applied Biosystems). Real time PCR protocols were performed as described previously[22]. Briefly, the PCR reaction mixture contained 2 mmol/L MgCl2, 20 pmol of both PCR primers, 1 pmol of each hybridization probe, 2 µL of LightCycler. DNA master hybridization mix (Roche, Germany) and 2 µL of reverse-transcribed samples in a final volume of 25 µL. The PCR conditions were set at 50 °C for 2 min, 95 °C for 10 min followed by 40 cycles of 95 °C for 30 s and 60 °C for 1 min. Additional reactions were performed on known dilutions of rat IL-1β cDNA as a PCR template to construct a standard curve relating threshold cycle to template copy number.
The sample copy numbers were calculated using the LightCycler analysis software (Roche, Mannheim, Germany). The LightCycler software system analyzed the spectral data collected at the end of the annealing phase of each cycle and plotted fluorescence intensity versus cycle number. The cDNA standard curve was generated by the LightCycler. The Cts value against the logarithm of the calculated initial copy numbers was plotted by the Software system. The unknown initial sample copy numbers were then calculated automatically from their Cts, as compared to the cDNA standard curve.
Taq polymerase primer and probe sets for IL-1β were designed from sequences in the GenBank database of NCBI on DNAman. GenBank is provided in the public domain by the National Center for Biotechnology Information, Bethesda, MD (http://www.ncbi.nlm.nih.gov/Genbank). The primer and probe sequences used were as follows: IL-1β forward primer: 5'-CTGCACTGCAGGCTTCGA-3'; reverse primer: 5'-TGGA-GAGCTTTCAGCTCACATG-3'; Taqman probe: 5'-FAM-ATGAACAACAAAAATGCCTCGTGCTGTCTGTAMRA-3'.
Western blot analysis At 30 min, 6 h, 24 h, 72 h after injury (n=3 per time point), an 8-mm spinal cord segment was dissected 4 mm rostral and 4 mm caudal from the lesion epicenter from each group. The cord tissues were resuspended in a lysis buffer (Cell Signaling, Beverly, USA) and homogenized in a dounce homogezenizer on ice. Tissue homogenate samples were centrifuged at 14 000 rpm for 10 min at 4 °C, and the supernatant samples were stored at -30 °C. Protein concentrations in the cell lysates were determined by Bio-Rad (Richmond, CA) protein assay, as recommended by the manufacturer. For analyses by Western blot, 20 µL of each suspension sample was separated in 12% SDS-PAGE, and the proteins were transferred to polyvivylidene difluorid membranes. Blots were blocked with 5% nonfat dry milk in tris-buffered saline (TBS) for 1 h at room temperature and then the membranes were incubated with 1:200 diluted monoclonal rabbit anti-rat antibodies against IL-1β (Sigma) or caspase-3 (Sigma) overnight at 4 °C. The membranes were then processed with HRP-conjugated goat anti-rabbit secondary antibody (1:500; Sigma). Immunoreactive bands were quantified using image analysis software.
p38 MAPK enzymatic activity was measured using a p38 MAP Kinase Assay Kit (Cell Signaling), and the protocol was performed according to the manufacturer’s instructions. Briefly, 200 µL of protein content in clear supernatant was immunoprecipited using immobilized phospho-p38 MAP kinase (Thr180/Tyr182) monoclonal antibodies with gentle shaking overnight at 4 °C. The immunoprecipitate was washed twice with 500 µL ice-cold cell lysis buffer and twice with 500 µL ice-cold kinase buffer (25 mmol/L Tris (pH 7.5), 5 mmol/L β-glycerolphosphate, 2 mmol/L DTT, 0.1 mmol/L Na3VO4, and 10 mmol/L MgCl2) at 4 °C. The kinase reactions were conducted in the presence of 200 µmol/L ATP and 2 µg activating transcription factor-2 (ATF-2) fusion protein at 30 °C for 30 min. After the reaction had been terminated by the addition of 3×SDS sample buffer, the mixture was boiled for 5 min, followed by brief centrifugation. Phospho-p38MAPK was selectively measured by Western immuno-blotting as described previously[23], and then immunoreactive bands were quantified using image analysis software.
Immunohistochemistry At several time intervals after SCI (30 min, 6 h, 24 h, 72 h, 1 wk, 6 wk), animals were deeply anesthetized and perfused via cardiac puncture with 0.1 mol/L phosphate-buffered saline (PBS) 150 mL (pH 7.4) and subsequently with 4% paraformaldehyde in 0.1 mol/L PBS 250 mL. The spinal cord was carefully dissected, and an 8-mm segment containing the injured epicenter was dissected out and post-fixed by immersion in 4% paraformaldehyde for 3 h and then cryoprotected by immersion in 20% sucrose of PBS overnight. Tissue segments were embedded in OCT and frozen as described previously[24]. Free-floating transverse sections (10 µm) were cut with a cryostat.
Immunohistochemical staining was performed by the avidin-biotin complex method as described previously[25]. The concentration for rabbit anti-IL-1β and rabbit anti-caspase-3 primary antibodies was 1:200. The concentration for biotinylated anti-rabbit antibodies was 1:200. Positive immunoreactive labeling was observed qualitatively.
Terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate-biotin nick end labeling (TUNEL) Spinal cord specimens used for TUNEL staining were collected as described above for immunohistochemistry methods. The segment was embedded in paraffin as previously described[24] and 5 µm-thick serial transverse sections were collected. The sections were processed for TUNEL staining with an in site cell apoptosis detection kit, and the staining protocol was performed according to the manu-facturer’s instructions. The sections were counterstained with hematoxylin. The ratios of TUNEL positive cells to total cells were counted and analyzed by computer. All TUNEL analyses were carried out by investigators who were blind as to the experimental conditions.
Statistical analysis Data are expressed as mean±SD. Statistical significance was assessed with one-way ANOVA. The significant difference between pairs of groups was tested by post hoc analysis. P<0.05 was considered significant in all cases.
Results
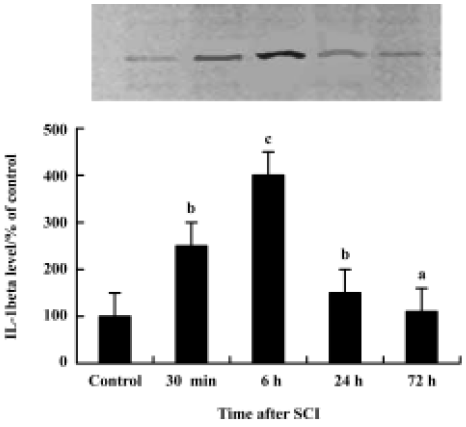
Rapid expression of IL-1β in spinal cord after SCI The temporal expression of IL-1β mRNA and protein at pre-defined time intervals (30 min, 6 h, 24 h, 72 h after SCI; n=3 for each time point) was examined. Absolute quantification real-time reverse PCR (Q-RT-PCR) analysis revealed that by 30 min after the contusion injury, IL-1β mRNA levels (6.75±0.42, log of the copy number of IL-1β cDNA) were significantly increased in the cords of injured rats relative to sham surgery control rats (4.98±0.49). IL-1β mRNA levels continued to rise, reaching peak levels at 6 h post-injury (6.97±0.75) (cP<0.01 vs control group), then falling by 24 h.
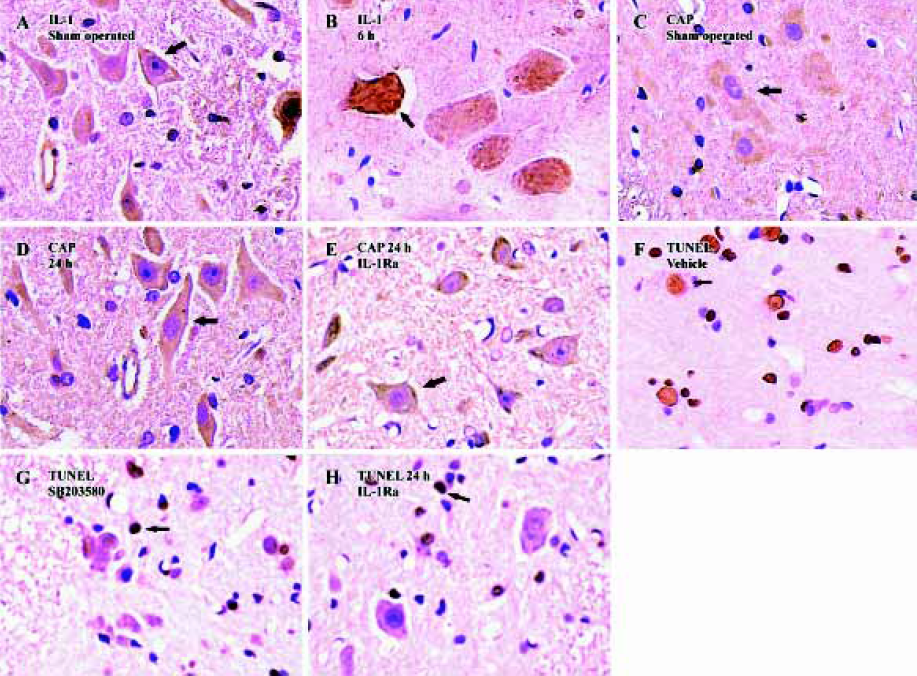
Western blot analysis also showed an increase in IL-1β levels following SCI (Figure 1). IL-1β protein was increased significantly by 30 min after the injury, reached peak levels by 6 h after the injury and declined thereafter. Both Western blot analysis (Figure 1) and immunohistochemistry examination (Figure 2A) revealed low levels of IL-1β protein in the sham controls. As early as 30 min after injury IL-1β immunoreactivity was observed in neurons within the gray matter in the lesion area, as well as in neurons located several microns, rostral and caudal of the lesion. The peak of immunoreactivity was observed 6 h after SCI (Figure 2B).
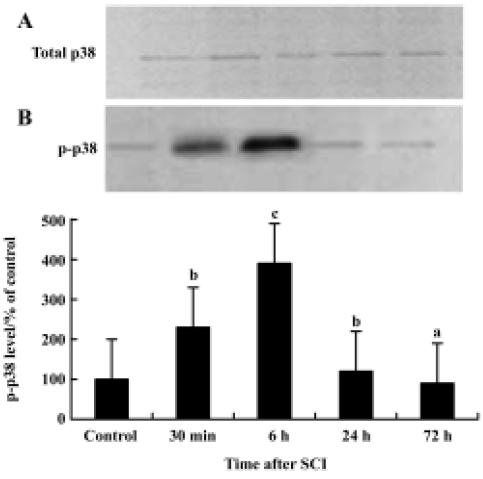
p38 MAPK activation in the spinal cord after injury Immunoprecipitation and Western blot examinations revealed that 6 h after SCI total p38 MAPK levels in the spinal cord had not changed after contusion (Figure 3A). P-p38 MAPK levels however were significantly increased following the injury. This increase was detectable at 30 min after contusion and reached a peak at 6 h. P-p38 MAP returned to basal levels by 72 h post-injury (Figure 3B). These data indicate that increases in p-p38 after SCI are caused by increased phosphorylation rather than by elevated substrate.
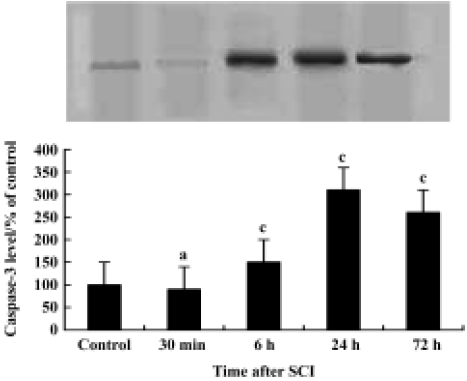
Induction of caspase-3 after injury The post-injury temporal profile of caspase-3 expression was examined by immunohistochemistry and Western blot analysis. Both injured and sham-operated spinal cords were evaluated for caspase-3 expression at four pre-defined time points (30 min, 6 h, 24 h, and 72 h after SCI; n=3 for each time point). As expected, little caspase-3 immunoreactivity was observed in sham-operated spinal cords (Figure 2C). Weak caspase-3 immunoreactivity was first detected in a few neurons within the gray matter of the lesioned area 6 h after injury. Intense caspase-3 immunoreactivity was seen in gray matter neurons 24 h after injury and decreased thereafter (Figure 2D). Western blot analyses also showed that caspase-3 expression was up-regulated after SCI. Caspase-3 protein was detectable by 6 h, peaked 24 h post-injury and decreased thereafter (Figure 4).
SCI-induced p38 MAPK activation is mediated by IL-1β To test whether IL-1β plays a role in p38 MAPK activation after SCI, the IL-1 receptor antagonist, recombinant rat IL-1ra, was given after SCI. Western blot data showed an increase in p-p38 in spinal cord 6 h after SCI. Treatment with IL-1Ra markedly inhibited the injury-induced activation of p38 MAPK. These data suggest that p38 MAPK activation in spinal cord after SCI is mediated by IL-1β (Figure 5).
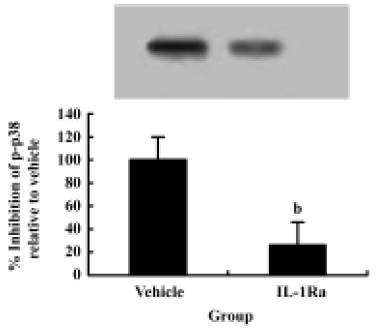
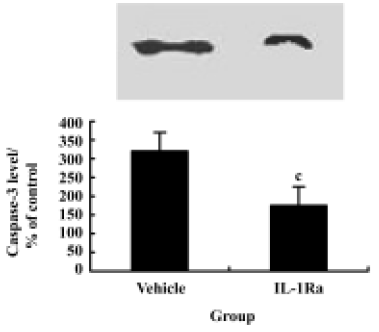
Inhibition of caspase-3 expression and reduction of apoptosis by IL-1Ra after injury To demonstrate whether the IL-1β induced expression of caspase-3 and the subsequent increased apoptosis of neurons after contusion injury, we compared both expression of caspase-3 and the number of apoptotic cells in the presence or absence of IL-1Ra after SCI. As shown in Figure 6, the induction of caspase-3 protein was inhibited significantly by IL-1Ra treatment. Few TUNEL-positive cells were initially present in the lesion area within 6 h of injury. The greatest numbers of TUNEL-positive cells were observed 24 h after injury. Treatment with IL-1Ra markedly reduced the number of TUNEL-positive cells in the cord relative to vehicle controls at 24 h after injury (Figure 2F, 2G).
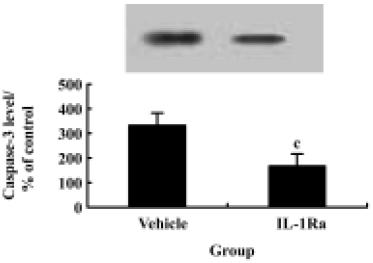
p38 MAPK inhibition reduces neuron apoptosis and caspase-3 expression after SCI Treatment of the injured spinal cord with SB203580 resulted in a marked reduction in the number of TUNEL-positive cells as compared to that found in vehicle controls (Figure 2H). Notably, at 24 h post-injury the number of TUNEL-positive cells was reduced by 50% with the SB203580 treatment. The induction of caspase-3 protein was also inhibited significantly by treatment with SB203580 (Figure 7).
Discussion
Injury to the spinal cord induces a complex cascade of cellular reactions at the local lesion area that results in inflammation and secondary cell death[26]. IL-1β is a major mediator of inflammation and may play an important role in the consequences of traumatic spinal cord injury. Numerous studies have confirmed an increase in IL-1β expression after SCI[6–8]. In the present study, the expression of IL-1β and its mRNA was assessed in the rat spinal cord following a standardized contusion injury. IL-1β mRNA levels, as measured by quantitative RT-PCR, were significantly increased in the lesion area by 30 min after SCI, peaked at 6 h and returned to normal at 72 h post-injury. IL-1β protein levels, as measured by Western blot and immunohistochemical staining, were increased significantly in the lesion area by 30 min post-injury, peaked at 6 h and then remained significantly higher than normal through at least 3 d post-injury. These findings, along with the findings by others[6–8], suggest that the increased IL-1β mRNA and protein levels are an early and local response at the lesion site that could trigger other, later, responses to traumatic SCI.
The function of IL-1β remains unclear. Some evidence indicates that IL-1β may exert beneficial effects on the CNS after injury such that IL-1β and its type 1 receptor may be involved in the maintenance of cell survival rather than induction of neuronal death. Direct application of inflammatory cytokines with SCI has been shown to reduce tissue loss[27]. On the contrary, however, findings of several studies are consistent with the view that an increased level of IL-1β after injury is adverse. Namely, increased IL-1β in spinal cord has been associated with neuronal apoptosis[4,9]. Direct injection of IL-1β into the spinal cord enhances vascular permeability and produces lymphocyte recruitment[28]. As such, an early and rapid production of IL-1β after injury appears to be deleterious to the CNS by triggering apoptosis. It may be that whether one sees enhanced cell survival or augmented apoptosis depends of the dose of IL-1β present in the cord.
The IL-1β expression peak (6 h post-injury) occurred when few TUNEL-positive apoptotic cells were present in the lesion area. The number of observed apoptotic cells peaked much later, 24 h after the contusion injury. IL-1β receptor antagonism with IL-1Ra significantly reduced the number of apoptotic cells present after injury. These data suggest that IL-1β plays a causal role in neuronal apoptosis in the early phase of SCI. However, the intracellular signaling events initiated by IL-1β that lead to apoptotic cell death remain largely unknown.
A number of studies have provided evidence that cell death from moderate traumatic spinal cord injury is regulated, in part, by apoptosis that involves the caspase family of cysteine proteases. A recent study demonstrated that spinal cord injury rapidly increased activation of the effector caspase, caspase-3. The caspase-3 message was present and upregulated several fold after traumatic spinal cord injury[29]. Moreover, upstream and downstream components of the caspase-3 apoptotic pathway are activated after traumatic spinal cord injury in rats. These effects occur early in neurons in the injury site and hours to days later in oligodendrocytes adjacent to and distant from the injury site[11]. Nesic O et al[9] showed that downstream components of the caspase-3 apoptotic pathway were activated after SCI in the rat. Furthermore caspase inhibition reduced post-traumatic lesion size and improved motor performance[10], indicating that caspase-3 is a potential mediator of apoptosis following CNS injury[10,11]. However it has not been shown definitively whether IL-1β induces apoptosis after SCI by activating caspase-3. The present Western blot and immunohistochemistry analyses results showed that caspase-3 expression was up-regulated in a relatively small number of neurons within the gray matter of the lesioned area at 6 h after SCI. Intense caspase-3 immunoreactivity was seen in gray matter neurons by 24 h after injury. Furthermore, IL-1 receptor antagonist treatment after SCI attenuated injury-induced upregulation of caspase-3 gene expression and resulted in fewer TUNEL-positive cells. Thus, these results are consistent with the hypothesis that IL-1β-induced apoptosis is mediated via induction of caspase-3 expression following SCI.
The MAPKs are a family of signaling molecules that transduce extracellular stimuli into intracellular responses in a wide variety of circumstances[15,30]. p38 MAPK is a key component in stress-induced signal transduction pathways. To test whether p38 MAPK signaling is involved in IL-1β induction of apoptosis, we examined the expression of p38 MAPK after SCI with immunoprecipitation and Western blot techniques. Increased levels of phospho-p38 MAPK were observed following SCI. Intrathecal injection of IL-1Ra attenuated the expression of caspase-3, and reduced the number of TUNEL-positive cells. In addition, treating the injured spinal cord with the MAPK inhibitor SB203580 resulted in a marked reduction in the expression of caspase-3 and the number of TUNEL-positive cells in the cord as compared to that observed in vehicle controls. These results are consistent with the hypothesis that IL-1β-induced apoptosis is mediated in part by p38 MAPK activation following SCI, and further that the apoptotic signal may then subsequently be mediated by caspase-3.
In conclusion, our study provides evidence that IL-1β serves as an external apoptosis triggering signal in the spinal cord after injury. Our results further suggest that the apoptotic cell death induced by IL-1β is mediated, in part, by phosphorylation of p38 MAPK and subsequent activation of the apoptotic gene caspase-3. These findings suggest that inhibition of p38 MAPK after SCI (ie by SB203580) may prevent neuronal cell death after SCI.
References
- Beattie MS, Hermann GE, Rogers RC, Bresnahan JC. Cell death in models of spinal cord injury. Prog Brain Res 2002;137:37-47.
- Emery E, Aldana P, Bunge MB, Puckett W, Srinivasan A, Keane RW, et al. Apoptosis after traumatic human spinal cord injury. J Neurosurg 1998;89:911-20.
- Fankhauser C, Friedlander RM, Gagliardini V. Prevention of nuclear localization of activated caspases correlates with inhibition of apoptosis. Apoptosis 2000;5:117-32.
- Ehrlich LC, Peterson PK, Hu S. Interleukin (IL)-1beta-mediated apoptosis of human astrocytes. Neuroreport 1999;10:1849-52.
- Takahashi JL, Giuliani F, Power C, Imai Y, Yong VW. Interleukin-1beta promotes oligodendrocyte death through glutamate excitotoxicity. Ann Neurol 2003;53:588-95.
- Hayashi M, Ueyama T, Nemoto K, Tamaki T, Senba E. Sequential mRNA expression for immediate early genes, cytokines, and neurotrophins in spinal cord injury. J Neurotrama 2000;17:203-18.
- Pan JZ, Ni L, Sodhi A, Aguanno A, Young W, Hart RP. Cytokine activity contributes to induction of inflammatory cytokine mRNAs in spinal cord following contusion. J Neurosci Res 2002;68:315-22.
- Wang CX, Olschowka JA, Wrathall JR. Increase of interleukin-1beta mRNA and protein in the spinal cord following experimental traumatic injury in the rat. Brain Res 1997;759:190-6.
- Nesic O, Xu GY, McAdoo D, High KW, Hulsebosch C, Perez-Pol R. IL-1 receptor antagonist prevents apoptosis and caspase-3 activation after spinal cord injury. J Neurotrauma 2001;18:947-56.
- Li M, Ona VO, Chen M, Kaul M, Tenneti L, Zhang X, et al. Functional role and therapeutic implications of neuronal caspase-1 and -3 in a mouse model of traumatic spinal cord injury. Neuroscience 2000;99:333-42.
- Springer JE, Azbill RD, Knapp PE. Activation of the caspase-3 apoptotic cascade in traumatic spinal cord injury. Nat Med 1999;5:943-6.
- Lynch AM, Lynch MA. The age-related increase in IL-1 type I receptor in rat hippocampus is coupled with an increase in caspase-3 activation. Eur J Neurosci 2002;15:1779-88.
- Rothwell NJ, Luheshi GN. Interleukin 1 in the brain: biology, pathology and therapeutic target. Trends Neurosci 2000;23:618-25.
- Maas JW Jr, Horstmann S, Borasio GD, Anneser JM, Shooter EM, Kahle PJ. Apoptosis of central and peripheral neurons can be prevented with cyclin-dependent kinase/mitogen-activated protein kinase inhibitors. J Neurochem 1998;70:1401-10.
- Widmann C, Gibson S, Jarpe MB, Johnson GL. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol Rev 1999;79:143-80.
- Aine K, Emily V, Yvonne N, Marcella B, Claire B, Christine EL, et al. Activation of p38 plays a pivotal role in the inhibitory effect of lipopolysaccharide and interleukin-1β on long term potentiation in rat dentate gyrus. J Biol Chem 2003;278:19453-62.
- Cuenda A, Rouse J, Doza YN, Meier R, Cohen P, Gallagher TF, et al. SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett 1995;364:229-33.
- Zheng RM, Zou CJ. ZHU SG. p38 MAPK mediates cardiovascular and behavioral responses induced by central IL-1β and footshock in conscious rats. Acta Pharmacol Sin 2004;25:266-70.
- Basso DM, Beattie MS, Bresnahan JC. Graded histological and locomotor outcomes after spinal cord contusion using the NYU weight-drop device versus transection. Exp Neurol 1996;139:244-56.
- Mulcahy NJ, Ross J, Rothwell NJ, Loddick SA. Delayed administration of interleukin-1 receptor antagonist protects against transient cerebral ischaemia in the rat. Br J Pharmacol 2003;140:471-6.
- Horiuchi H, Ogata T, Morino T, Chuai M, Yamamoto H. Continuous intrathecal infusion of SB203580, a selective inhibitor of p38 mitogen-activated protein kinase, reduces the damage of hind-limb function after thoracic spinal cord injury in rat. Neurosci Res 2003;47:209-17.
- Hernandez-Rodriguez J, Segarra M, Vilardell C, Sanchez M, Garcia-Martinez A, Esteban MJ, et al. Tissue production of pro-inflammatory cytokines (IL-1beta, TNFalpha and IL-6) correlates with the intensity of the systemic inflammatory response and with corticosteroid requirements in giant-cell arteritis. Rheumatology (Oxford) 2004;43:294-301.
- Jin SX, Zhuang ZY, Woolf CJ, Ji RR. p38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. J Neurosci 2003;23:4017-22.
- Lee YB, Yune TY, Baik SY, Shin YH, Du S, Rhim H, et al. Role of tumor necrosis factor-α in neuronal and glial apoptosis after spinal cord injury. Exp Neurol 2000;166:190-5.
- Tomlinson A, Appleton I, Moore AR, Gilroy DW, Willis D, Mitchell JA, et al. Cyclo-oxygenase and nitric oxide synthase isoforms in rat carrageenin-induced pleurisy. Br J Pharmacol 1994;113:693-8.
- Bartholdi D, Schwab ME. Expression of pro-inflammatory cytokine and chemokine mRNA upon experimental spinal cord injury in mouse: an in situ hybridization study. Eur J Neurosci 1997;9:1422-38.
- Klusman I, Schwab ME. Effect of pro-inflammatory cytokines in experimental spinal cord injury. Brain Res 1997;762:173-84.
- Schnell L, Fearn S, Schwab ME, Perry VH, Anthony DC. Cytokine-induced acute inflammation in the brain and spinal cord. J Neuropathol Exp Neurol 1999;58:245-54.
- Citron BA, Arnold PM, Sebastian C, Qin F, Malladi S, Ameenuddin S, et al. Rapid upregulation of caspase-3 in rat spinal cord after injury: mRNA, protein, and cellular localization correlates with apoptotic cell death. Exp Neurol 2000;166:213-26.
- Lee JC, Laydon JT, McDonnell PC, Gallagher TF, Kumar S, Green D, et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature 1994;372:739.