
Is COX-2 a perpetrator or a protector? Selective COX-2 inhibitors remain controversial1
Introduction
Cyclooxygenase (COX) is a key enzyme in the conversion of polyunsaturated fatty acids and arachidonic acid to prostaglandin (PG) H2, which is further converted into various prostanoids (PGs, prostacyclins and thromboxanes). Our knowledge about COX constantly changes, but basically COX-1 is constitutively expressed in most tissues, where it synthesizes physiological amounts of postaglandins. COX-2, on the other hand, is normally expressed in activated macrophages and becomes strongly upregulated after exposure to growth factors or inflammatory stimuli, and is elevated in malignant cells to promote angiogenesis in tumors[1–4]. It has been long believed that many of the side effects of nonsteroidal anti-inflammatory drugs (NSAIDs), for examples, gastrointestinal ulceration and bleeding, and platelet dysfunction, are due to the suppression of COX-1-derived prostanoids, whereas the inhibition of COX-2-dependent prostaglandin synthesis accounts for the anti-inflammatory, analgesic and antipyretic effects. Consequently, it has been hypothesized that specific inhibition of COX-2 might have therapeutic actions similar to those of NSAIDs, but without the unwanted side effects. This was the fundamental rationale for the development of selective inhibitors of the COX-2 enzyme as a new class of anti-inflammatory and analgesic agent with improved gastrointestinal tolerability. However, the potential of this new class of drugs has not been realized, even though the rationale underpinning their use is likely to be correct.
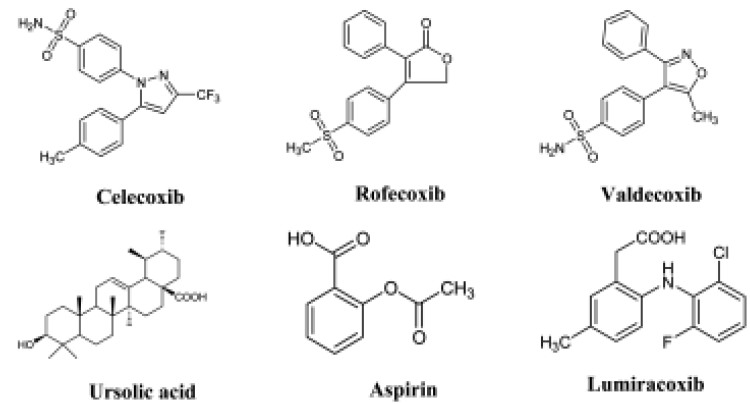
COX-2 was discovered in the early 1990s, and a large amount of effort has been expended since then by the pharmaceutical industry in the search for COX-2 inhibitors. In 2000, COX-2 inhibitors were involved in clinical trials for the treatment of colon and rectal cancers because COX-2 overexpression had been found in malignant tumor samples. However, at this point, the risk of heart attack and stroke emerged, and this is possibly because COX-2 is physiologically expressed in the glomeruli and cortex, and COX-2 may also have an anti-inflammatory role[5]. Recently, a number of events regarding COX-2 inhibitors have attracted the attention of the media. Rofecoxib (Vioxx, Figure 1) was withdrawn by Merck & Co in September 2004 following the finding from the Vioxx Gastrointestinal Outcomes Research (VIGOR) trial for colon and rectal cancers that there were severe risks for the heart. Similar claims were made when a COX-2 inhibitor was used in the adenoma prevention with celecoxib (APC) trial, which was eventually suspended in December 2004 because analysis by an independent Data Safety and Monitoring Board showed that there was a risk of fatal and non-fatal major cardiovascular disease (CVD)[6]. On February 20, 2005, a US Food and Drug Agency (FDA) advisory panel unanimously concluded that all classes of COX-2 inhibitors increase a person’s risk of heart attack and stroke, and recommended that, despite the risks, drugs including Vioxx should remain on the market but be accompanied by strong warnings. Following the FDA’s decision, the European Union and countries in which COX-2 inhibitors were used imposed similar restrictions. Because the drugs are now “tainted”, physicians are reluctant to prescribe them, and patients do not want to risk the health of their heart to ease pain, the sales of Celebrex, valdecoxib (Figure 1) and rofecoxib have dropped dramatically. As COX-2 inhibitors have become controversial, many pharmaceutical companies are having to renew their research efforts in the search for new effective anti-inflammatory drugs to treat arthritis and other inflammatory diseases[7].
Regulation of COX-2 expression during inflammation
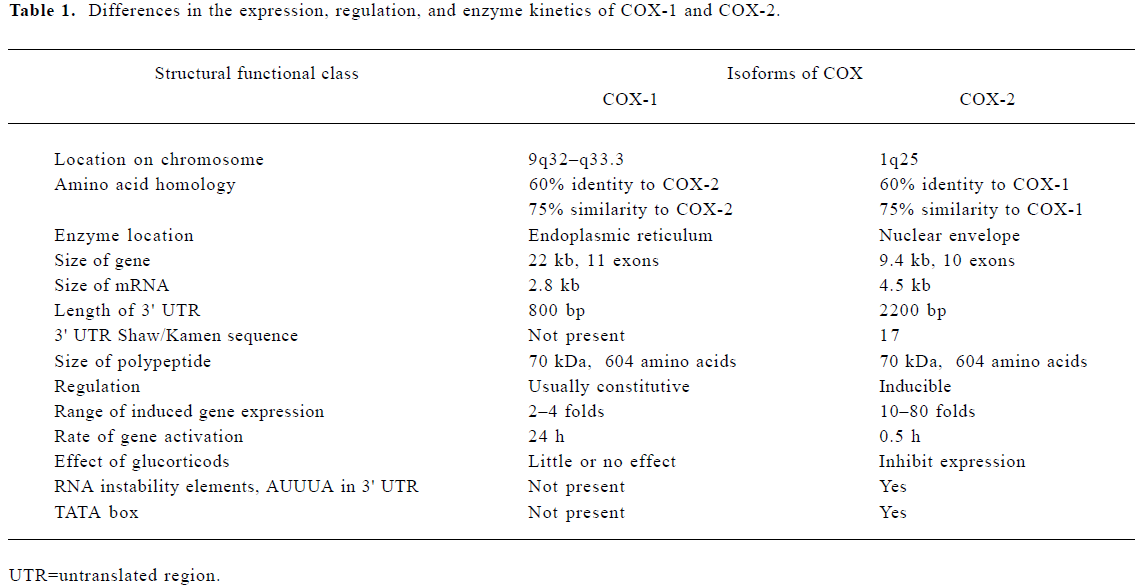
One of the main reasons for COX-2 attracting pharmaceutical interest is its strikingly different method of gene regulation from COX-1. The genes of COX-1 and COX-2 are located in human chromosomes 9 and 1, respectively, with COX-1 lacking a TATA box[8]. The promoter of the immediate-early gene COX-2 contains a TATA box and binding sites for several transcription factors that are different from those of COX-1 (Table 1). The expression of COX-2 is regulated by a broad spectrum of mediators involved in inflammation. In general, lipopolysaccharides and proinflammatory cytokines, for example, interleukin- (IL)-1β, tumor necrosis factor (TNF), and growth factors, induce COX-2; whereas IL-4, IL-13 and the anti-inflammatory cytokine IL-10 inhibit the expression of the enzyme[9,10]. A recent study has shown that there is a positive feedback loop in COX-2-dependent prostaglandin production at the sites of inflammation. In particular, a COX-2 product, PGE2, upregulates COX-2 expression by virtue of its cAMP-elevating capacity in human blood monocytes[11]. COX-2 is also regulated at the post-transcriptional level. Recently, a 3'-untranslated region of the COX-2 mRNA has been shown to contain multiple copies of adenylate- and uridylate-rich elements (AREs) that may confer post-transcriptional control of COX-2 expression by acting as mRNA instability determinants or as translation inhibitory elements. Loss of this post-transcriptional regulation of COX-2 through the mutation of proteins that specifically interact with the COX-2 ARE may lead to COX-2 overexpression, and this process has been proposed as a crucial factor in colon carcinogenesis[12,13]. The sustained increase of COX-2 expression is apparently due (at least in part) to IL-1A increasing the stability of COX-2 mRNA. This type of regulatory mechanism may play an important role in chronic inflammatory conditions.
Full table
Development of COX-2 inhibitors
Both COX-1 and COX-2 produce prostaglandin H2, which is further converted to different prostanoids, including PGD2, PGE2, PGF2alpha, PGI2 and thromboxane A2. COX-2-dependent prostaglandins have been implicated in various physiological events, including male fertility, menstruation, ovulation, pregnancy, implantation, and in the pathological action of different inflammatory and neoplastic diseases, notably arthritis and cancer. In the US, one-third of the adult population, or approximately 70 million people, are affected by arthritis or chronic joint symptoms, making arthritis one of the most prevalent diseases in that country. Onset of the disease starts at around the age of 30 years, and women have a higher incidence than men. Arthritis is the leading cause of disability in the US, accounting for 17.5% of all those on disability pensions. Thus there is an urgent need for the development of COX-2 inhibitors, and the development of suitable drugs would also be associated with large profits for pharmaceutical companies.
Inhibition of cyclooxygenases with aspirin can be traced back to ancient times. Chinese and Greek healers used willow bark that contained salicylic acid before AC aspirin, a modified form of acetylsalicylic acid developed by a German pharmacological chemist in 1898, was accepted as a safe and effective remedy for backache in 1903, for headache in 1923, and for arthritis in 1933 worldwidely. The mechanism by which aspirin exerts analgesic and antipyretic effects was found to be the inhibition of cyclooxygenase-dependent prostaglandins in 1971[14]. In the 1980s, aspirin was suggested to reduce the risk of heart attack by suppressing the production of prostacyclin in platelets[15]. Aspirin also slows the development of cataracts and reduces migraine attacks. However, gastrointestinal (GI) toxicity induced by aspirin and other NSAIDs is among the most common serious adverse drug events in the industrialized world[16]. Gastroduodenal ulcers can be found by endoscopy in 10% to 20% of patients who take NSAIDs on a regular basis. For decades, it has been a dream of pharmacologists to overcome this issue.
In the early 1990s, cyclooxygenase was found to exist in two distinct isoforms[1,2]. It was then very rapidly found that the COX-1 isoform is located in all tissues except red blood cells, and is constitutively expressed in the stomach, kidneys, platelets, and endothelial cells under normal physiological conditions. In contrast, COX-2 is induced by proinflammatory substances, such as lipopolysaccharides, TNF-alpha, IL-1, platelet-derived growth factor (PDGF), and other growth factors. COX-2 is involved in inflammation, for example in macrophages, monocytes and synoviocytes that emerge in acute and chronic inflammatory states. COX-2 therefore became a very attractive pharmaceutical target, even though it was later found that COX-2 is constitutively expressed in pancreatic beta-cells[17], at the distal end of the vas deferens[18], and also functionally and physiologically expressed in the kidney glomeruli and cortex[19]. COX-2 was quickly crystallized and the structure was resolved by X-ray diffraction crystallography, which is a crucial step in drug design. The techniques of investigating the docking between small molecules and determining the structure of COX-2 were able to be carried out using different platforms. These approaches help to understand the mechanisms involved in protein–ligand binding in general, as well as helping to understand the details of the interactions in a specific protein or protein-ligand complex of interest[20]. Celecoxib was the first drug that was chemically designed by computer, and was exceptionally quickly approved for release by the FDA in December 1998. Rofecoxib and valdecoxib subsequently entered the market later. The makers of a new product containing lumiracoxib (Novartis) are now in the process of applying for a license. Lumiracoxib is claimed to reduce the from cancer. Prescriptions for COX-2-selective inhibitors represented up 61% of total prescriptions for NSAIDs in 2001 and 2002. In 2003, celecoxib was the ninth best-selling drug in the US, with sales of US$2.6 billion according to Forbes. Another impetus for COX-2 inhibitor development is for cancer chemotherapy. In one study, COX-2 knockout mice showed a reduced incidence of tumors[21], and COX-2 is overexpress-ed in many kinds of cancers, including colon, prostate and lung cancer. The function of COX-2 inhibitors that is relevant for cancer treatment is that they also induce apoptosis.
Mechanism of action of COX-2-dependent PGE2 in pain perception
Inflammation causes an increased synthesis of COX-2-dependent PGs, which sensitize peripheral nociceptor terminals and produce localized pain hypersensitivity. A significant portion of these nociceptors are not activated by physiological stimuli such as gentle pressure or temperature. PGs regulate the sensitivity of so-called polymodal nociceptors that are present in nearly all tissues. PGE2 and other inflammatory mediators facilitate the activation of tetrodotoxin (TTX)-resistant Na+ channels in dorsal root ganglion neurons. These small dorsal root ganglion neurons are unmyelinated nerve fibers, conducting nociceptive stimuli. During inflammatory states, PGE2 receptor E1 may significantly increase the excitability of nociceptive nerve fibers, thereby contributing to the activation of “sleeping” nociceptors, and also act in the central nervous system to produce hyperalgesia. COX-2 inhibitors act primarily in the dorsal horn to cause analgesia. The sensation of pain is assembled in the cortex. COX-2 is expressed constitutively in the dorsal horn of the spinal cord, and becomes upregulated briefly after a trauma, such as damage to a limb, in the corresponding sensory segments of the spinal cord. The induction of spinal cord COX-2-dependent PGs facilitates transmission of the nociceptive input[22]. Similar to in vitro IL-1-induced COX-2 expression, IL-1β was demonstrated to be the major inducer of COX-2 upregulation in the central nervous system. Accordingly, intraspinal administration of an interleukin-converting enzyme or COX-2 inhibitor is accompanied by decreases in both inflammation-induced central PGE2 levels and mechanical hyperalgesia[23,24]. For analgesia alone, opioid drugs are more effective in moderate-to-severe pain than NSAIDs, and possess other significant advantages including titratability, reversibility and lack of prostaglandin-associated side effects. Cocktails of weak opioids and other analgesic agents, such as tramadol plus acetaminophen, have been shown to have some merits[25].
After a debate lasting 30 years, the mechanism by which acetaminophen (commercial name Tylenol) exerts its analgesic and antipyretic action has also been elucidated by cyclooxygenase isoform studies. In contrast to the acidic antipyretic selective COX-2 inhibitors, due to the non-acidic chemistry of acetaminophen, the drug reaches higher concentrations within the central nervous system as compared with the acidic antipyretic analgesics (eg, aspirin and indomethacin) that accumulate in peripheral compartments with acidic extracellular pH (eg, inflamed tissue). Acetaminophen may inhibit nociception-induced spinal prostaglandin synthesis, and hydroperoxide may confer cellular selectivity on acetaminophen’s inhibition of COX-2 activity[26]. In addition, acetaminophen has been suggested to inhibit a newly discovered COX isoform, derived from the same gene as COX-1 and referred to as COX-1b or COX-3. Aceta-minophen’s role in analgesia and hypothermia is due to the inhibition of COX-3, which is more dominantly involved in fever in children[27].
Synthetic COX-2 selective inhibitor with neph-rotoxicity and cardiovascular disease
COX-2 immediately became a drug target after it was characterized as an inducible isoform in the early 1990s. Several crystal structures of COX-2 were quickly solved through the crystallization of the protein in different laboratories within a few years. In 1996, Searle improved the model and built up a structure with a high resolution of 2.15 Å. It was found that one of the enzymatic channels in COX-2 is wider than that of COX-1, and COX-2 has an open side pocket where the sulfonamide group of the selective inhibitors can bind. Human COX-2 has four amino acids that are different from COX-1 in the region corresponding to the side pocket. The most important difference is at position 523, where COX-1 has a bulky isoleucine and COX-2 has a valine that takes up less space[28]. The formation of five different hydrogen bonds between the selective inhibitors and the pocket of COX-2 is a strong and time dependent binding reaction, and site-directed mutations in the pocket region resulted in weaker binding and fewer hydrogen bonds[20,29]. The selectivity of COX-2 IC50/COX-1 IC50 is 0.003 (or selective fold is 375:1) for celecoxib, 0.0026 for refecoxib, and 166 for aspirin. The high selectivity of celecoxib is due to the phenylsulfonamide moiety, which binds in a pocket that is more restricted in COX-1 and is unoccupied in complexes of COX-2 with non-selective inhibitors. This pocket branches off from the main channel that leads to the cyclooxygenase active site, and is more accessible in COX-2, primarily because of the previously mentioned substitution of isoleucine at position 523 in COX-2 to valine in COX-1. Aspirin mainly inhibits COX-1 irreversibly because it blocks the channel for accessing the substrate by permanently acetylating the serine of the cyclooxygenase enzymes. Celecoxib’s reversibility and selectivity provided a new insight into drug design approaches for the pharmaceutical industry.
Nevertheless, the COX-2 inhibitors are associated with a number of potential pitfalls, which can be even more lethal than the side effects of other non-selective NSAIDs. These pitfalls arise from the fact that COX-2 induction is not exclusively associated with the onset of an inflammatory reaction. In fact, COX-2 can also be expressed chronically, and is seen during the resolution of inflammation and in areas of wound-healing[5]. Through studies of COX-2 knockout mice, COX-2 inhibition has been directly linked to several adverse outcomes, including reproductive failure, and reduced renin expression in the kidney. Renin is an active enzyme that converts angiotensinogen into angiotensin, which maintains or modulates the salt metabolism. It has been shown that renin expression is decisively regulated by COX-2 in kidney, especially in the abnormal intake of NaCl[30]. COX-2-deficient mice also develop lung fibrogenesis[31], which is a common attribute of lung cancer in humans. The application of COX-2-selective inhibitors during these periods has been shown to be deleterious, so that resolution of inflammation and gastric ulcer healing are actually delayed and, in some patients, ulcers have even been found to progress further to perforation. The suggestion has now been made that, in these situations, COX-2 may actually help resolve the pathology, perhaps by generating an alternative series of PGs, such as the cyclopentenone PGs. The finding that these PGs can affect proteins by direct chemical modifications as well as by having their own receptor families has rekindled debate on the deleterious and beneficial effects of COX-2-dependent prostanoids, and on the implications of inhibiting the production of these mediators in the body[32].
COX-2-synthesized PGs have important roles in the modulation of renal physiology. A number of pooled analyses have suggested the possibility of an increased risk of CVD, particularly acute myocardial infarction, because of a blunted PG response due to COX-2 inhibition in the kidney and blood vessels, which may depend on the individual to a certain extent. Many reports have stated that COX-2 inhibitors have the same nephrotoxicity as non-selective NSAIDs. PGs have a counterbalancing effect because they modulate the local actions of systemic and locally produced vasoconstrictor hormones. For this role, a variety of PGs with diverse biological effects are produced and metabolized by the kidney[33]. The major ones, synthesized at distinct anatomical locations within the renal parenchyma, include PGI2, PGE2, thromboxane A2, and PGF2. Of these, PGI2 (produced most abundantly in the renal cortex by cortical glomeruli and arterioles) and PGE2 (synthesized in the juxtamedullary glomeruli, medullary interstitial cells, and the medullary portion of the collecting duct) are physiologically predominant[34,35]. It used to be believed that the only side effects of COX-2 inhibitors were toxicity in the kidney. But now COX-2 has been found to be constitutively expressed in the endothelial cells of the arteries, arterioles, lung and brain, in addition to its highly constitutive expression in the vas deferens and beta-cells of the pancreas. COX-2 expression in the cortical thick ascending limb of the loop of Henle (medullary rays and macula densa) affects chloride ion pumps. In the medulla, COX-2 expression was detected in the endothelial lining of the vasa recta in 52 cases and in the collecting ducts in 5 cases. These data demonstrate significant constitutive expression of COX-2 in normal kidney and underscore the need for caution in the use of COX-2 selective inhibitors, especially on a long-term basis. All in all, there is not much remaining of the original dogma that COX-1 is the housekeeping good guy and COX-2 is the perpetrator that mediates inflammation and damage. These finding are consistent with recent clinical data showing that COX-2 inhibitors are associated with CVD risk[36,37].
Naturally derived COX-2 inhibitors for anti-inflammation, and alternative targets for painkilling
Given that salicylic acid (later developed to aspirin) was originally extracted from the bark of the willow tree, there is a definite possibility of also finding COX-2 inhibitors in nature. In fact, there are some already known COX-2 favorable inhibitors of natural origin, and some of them have been used as natural modulators of inflammation in adjuvant chemotherapy. A recent review details a number of natural COX-2 inhibitors with different chemical structures and different modes of action[38]. It has been reported that ginger, curcumin, thunder god vine, plantago, and others contain COX-2 inhibitors. Some of these compounds not only inhibit COX-2, but are also antioxidants, for example resveratrol, which is found in the skin of grapes and inhibits the transcription or post-translation of the COX-2 gene[39]. Small synthetic COX-2 inhibitors can be inserted into loops that are different from COX-1. But there are many different methods of inhibition, and in general, there is no correlation between inhibition and the similarity of the chemical structures. Some characterized fatty acid COX-2 inhibitors, such as linoleic acid (LA), alpha-linolenic acid (alpha-LNA), myristic acid and palmitic acid have been isolated from different plant roots or leaves, and have IC50 values for cyclooxygenase ranging from 3.9 to180 µmol/L. The compound alpha-LNA is one of the most selective towards COX-2, with COX-2/COX-1 ratios of 0.1[40,41].
In addition to fatty acids, steroid-like molecules are also attractive candidates for COX-2 inhibitors. A hexane structure of ursolic acid (Figure 1) has been investigated by bioactivity-directed fractionation. This triterpenoid had a COX-2 inhibitory effect, directly affecting the enzyme activity, with an IC50 value of 130 µmol/L and a COX-2/COX-1 selectivity ratio of 0.6. The structural isomer oleanolic acid is less active than ursolic acid, with an IC50 value of 295 µmol/L, but has a similar selectivity ratio (0.8). The direct inhibitory effect of ursolic acid and oleanolic acid on COX-2-catalyzed prostaglandin biosynthesis increased with preincubation, indicating a time-dependent inhibition. Interestingly, the effect on COX-1 was independent of preincubation time[42]. The problem with natural COX-2 inhibitors is the slower onset of relief in vivo. Effort needs to be directed towards screening natural anti-inflammatory/analgesic products that offer more effective inhibition, or stronger instantaneous relief of pain with no (or less) GI and cardiovascular risk. If this were to occur, then a natural COX-2 inhibitor, whether a single compound or a combination of multiple compounds, could be developed to provide gentle, effective and long-term relief.
Summary
Cyclooxygenases, particularly COX-2 inhibitors associated with CVD problems, have shown us that there are probably no absolute perpetrators and protectors in nature. However, selective COX-2 inhibitor studies and chemotherapy have extended our understanding of this system. All COX-2 inhibitors remain available for use in certain patients, such as those at risk of ulcers. COX-2 inhibitors can be explored further for cancer therapy, where because of their anti-angiogenic apoptosis-promoting properties, they might benefit those patients who are overexpressing COX-2. Rather than inhibitors, it is also possible to identify more COX-2 modulators, which could be found by screening small molecules from herbs or other natural sources. For simple painkilling, it is better to target end effectors. Screening for E1 receptor inhibitors is a new possibility because the analgesic actions of NSAIDs in inflammatory pain, especially visceral stimuli, are mediated to a significant degree by the inhibition of signaling through the E1 receptor. The absence of EP1 receptors results in structurally normal kidney, and does not result in a lowering of resting blood pressure[43].
References
- Xie WL, Chipman JG, Robertson DL, Erikson RL, Simmons DL. Expression of a mitogen-responsive gene encoding prostaglandin synthase is regulated by mRNA splicing. Proc Natl Acad Sci USA 1991;88:692-6.
- Fu JY, Masferr JL, Seibert K, Raz A, Needleman P. The induction and suppression of prostaglandin h2 synthase (cyclooxygenase) in human monocytes. J Biol Chem 1990;265:16737-40.
- Luo C, Laine JVO, Ylinen L, Teros T, Mäkinen M, Ristimäki A, et al. Expression of cyclooxygenase-2 in intestinal goblet cells of prediabetic non-obese diabetic mice. Acta Physiol Scand 2002;174:265-74.
- Luo C, Trauss L, Treng T, Ristimäki A, Santti R. Constant exprkession of cyclooxygenase-2 gene in the lower urinary tract and prostate of the male rats treated with estrogen. Z Naturforsch 2001;56C:455-63.
- Gilroy DW, Colville-Nash PR, Willis D, Chivers J, Paul-Clark MJ, Willoughby DA. Inducible cyclooxygenase may have anti-inflammatory properties. Nat Med 1999;5:621-2.
- Topol EJ. Arthritis Medicines and Cardiovascular Events: “House of Coxibs J Am Med Assoc 2005;293:366-8.
- Warner TD, Mitchell JA. Cyclooxygenases: new forms, new inhibitors, and lessons from the clinic. FASEB J 2004;18:790-804.
- Kraemer SA, Meade EA, DeWitt DL. Prostaglandin endoperoxide synthase gene structure: identification of the transcriptional start site and 5'-flanking regulatory sequences. Arch Biochem Biophys 1992;293:391-400.
- Colville-Nash PR, Gilroy DW. Potential adverse effects of cyclooxygenase-2 inhibition: evidence from animal models of inflammation. BioDrugs 2001;15:1-9.
- Onoe Y, Miyaura C, Kaminakayashiki T, Nagai Y, Noguchi K, Chen QR, et al. IL-13 and IL-4 inhibit bone resorption by suppressing cyclooxygenase-2-dependent prostaglandin synthesis in osteoblasts. J Immunol 1996;156:758-64.
- Hinz B, Brune K, Pahl A. Cyclooxygenase-2 expression in lipopolysaccharide-stimulated human monocytes is modulated by cyclic AMP, prostaglandin E2 and non-steroidal anti-inflammatory drugs. Biochem Biophys Res Commun 2000;278:790-6.
- Dixon DA, Kaplan CD, McIntyre TM, Zimmerman GA, Prescott SM. Post-transcriptional control of cyclooxygenase-2 gene expression. The role of the 3’- untranslated region. J Biol Chem 2000;275:11750-7.
- Ristimäki A, Honkanen N, Jankala H, Sipponen P, Harkonen M. Expression of cyclooxygenase-2 in human gastric carcinoma. Cancer Res 1997;57:1276-80.
- Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for the aspirin-like drugs. Nature 1971;231:232-5.
- Michiels JJ. Aspirin and platelet-lowering agents for the prevention of vascular complications in essential thrombocythemia. Clin Appl Thromb Hemost. 1999;5:247-51.
- Lichtenstein DR, Wolfe MM. COX-2 selective NSAID, new and improved? J Am Med Assoc 2000;284:1297-9.
- Luo C, Kallajoki M, Mäkinen M, Teros T, Ylinen L, Simell O. Distribution and contribution of cyclooxygenase (COX)-2 for diabetogenesis in NOD mouse model. Cell Tissue Res 2002;310:169-75.
- McKanna JA, Zhang MZ, Wang JL, Cheng H, Harris RC. Constitutive expression of cyclooxygenase-2 in rat vas deferens. Am J Physiol 1998;275:R227-33.
- Harris RC. Macula densa signaling, a potential role of cyclooxygenase-2 (COX-2)? Nephrol Dial Transplant 2000;15:1504-6.
- Luo C, Laaja P. Inhibitors of JAKs/STATs and the kinases, a possible new cluster of drugs. Drug Discov Today 2004;9:268-75.
- Tiano HF, Loftin CD, Akunda J, Lee CA, Spalding J, Sessoms A, et al. Deficiency of either cyclooxygenase (COX)-1 or COX-2 alters epidermal differentiation and reduces mouse skin tumorigenesis. Cancer Res 2002;62:3395-401.
- Baba H, Kohno T, Moore KA, Woolf CJ. Direct activation of rat spinal dorsal horn neurons by prostaglandin E2. J Neurosci 2001;21:1750-6.
- Samad TA, Moore KA, Sapirstein A, Billet S, Allchorne A, Poole S, et al. Interleukin-1b-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature 2001;410:471-5.
- Ma W, Eisenach JC. Morphological and pharmacological evidence for the role of peripheral prostaglandins in the pathogenesis of neuropathic pain. Eur J Neurosci 2002;15:1037-47.
- Ellman MH, Curran J. Trial of tramadol/acetaminophen tablets for osteoarthritis pain in subjects receiving a COX-2 nonsteroidal antiinflammatory drug. J Rheumatol 2005;32:568-8.
- Boutaud O, Aronoff DM, Richardson JH, Marnett LJ, Oates JA. Determinants of the cellular specificity of acetaminophen as an inhibitor of prostaglandin H2 synthases. Proc Natl Acad Sci USA 2002;99:7130-5.
- Botting R, Ayoub SS. COX-3 and the mechanism of action of paracetamol/acetaminophen. Prostaglandins Leukot Essent Fatty Acids 2005;72:85-7.
- van Ryn J, Trummlitz G, Pairet M. COX-2 selectivity and inflammatory processes. Curr Med Chem 2000;7:1145-61.
- Kurumbail RG, Stevens AM, Gierse JK, McDonald JJ, Stegeman RA, Pak JY, et al. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature 1997;385:555-62.
- Yang T, Endo Y, Huang YG, Smart A, Briggs JP, Schnermann J. Renin expression in COX-2-knockout mice on normal or low-salt diets. Am J Physiol Renal Physiol 2000;279:F819-25.
- Bonner JC, Rice AB, Ingram JL, Moomaw CR, Nyska A, Bradbury A, et al. Susceptibility of cyclooxygenase-2-deficient mice to pulmonary fibrogenesis. Am J Pathol 2002;161:459-70.
- Mukherjee D, Topol EJ. COX-2: where are we in 2003? Cardiovascular risk and COX-2 inhibitors. Arthritis Res Ther 2003;5:8-11.
- Adegboyega PA, Ololade O. Immunohistochemical expression of cyclooxygenase-2 in normal kidneys. Appl Immunohistochem Mol Morphol 2004;12:71-4.
- McAdam BF, Catella-Lawson F, Mardini IA, Kapoor S, Lawson JA, FitzGerald GA. Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: the human pharmacology of a selective inhibitor of COX-2. Proc Natl Acad Sci USA 1999;96:272-7.
- Catella-Lawson F, McAdam B, Morrison BW, Kapoor S, Kujubu D, Antes L, et al. Effects of specific inhibition of cyclooxygenase-2 on sodium balance, hemodynamics, and vasoactive eicosanoids. J Pharmacol Exp Ther 1999;289:735-41.
- Solomon SD, McMurray JJ, Pfeffer MA, Wittes J, Fowler R, Finn P, et al. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N Engl J Med 2005;352:1071-80.
- Bresalier RS, Sandler RS, Quan H, Bolognese JA, Oxenius B, Horgan K, et al. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med 2005;352:1092-102.
- Perera P, Ringbom T, Huss U, Vasänge M., Bohlin L. Search for natural products with effect on cyclooxygenase-2. In: Tringali C, editor. Bioactive Compounds from Natural Sources. London: Taylor & Francis; 2001. p433–72.
- Subbaramaiah K, Michaluart P, Chung WJ, Tanabe T, Telang N, Dannenberg AJ. Resveratrol inhibits cyclooxygenase-2 transcription in human mammary epithelial cells. Ann NY Acad Sci 1999;889:214-23.
- Ringbom T, Huss U, Stenholm A, Flock S, Skattebol L, Perera P, et al. COX-2 inhibitory effects of naturally occurring and modified fatty acids. J Nat Prod 2001;64:745-9.
- Arita M, Bianchini F, Aliberti J, Sher A, Chiang N, Hong S, et al. Stereochemical assignment, antiinflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. J Exp Med 2005;201:713-22.
- Ringbom T, Segura L, Noreen Y, Perera P, Bohlin L. Ursolic acid from Plantago major, a selective inhibitor of cyclooxygenase-2 catalyzed prostaglandin biosynthesis. J Nat Prod 1998;61:1212-5.
- Stock JL, Shinjo K, Burkhardt J, Roach M, Taniguchi K, Ishikawa T. The prostaglandin E2 EP1 receptor mediates pain perception and regulates blood pressure. J Clin Invest 2001;107:325-31.