
Ion channelopathy and hyperphosphorylation contributing to cardiac arrhythmias1
Introduction
Cardiac arrhythmias mostly occur in diseased hearts as a result of an abnormality in ion channels[1]. Diseased hearts with overt cardiac remodeling are at risk of developing severe and life-threatening arrhythmias. Patients suffering from sudden coronary death (SCD) were monitored with ECG to manifest ventricular tachyarrhythmias. Between 50% and 80% of deaths in patients suffering from congestive heart failure are caused by cardiac arrhythmias[2]. There have been significant advances in the molecular biological considerations of cardiac arrhythmias[3], however, there has been no success in suppressing ventricular tachyarrhythmias using antiarrhythmic agents[4,5]. Electronic devices and electrical defibrillators remain the first choice in controlling ventricular tachyarrhythmias.
Continuous effort has been made to search for new and effective antiarrhythmic agents for controlling life-threatening cardiac arrhythmias. The efficacy of antiarrhythmic agents to prevent cardiac sudden death was evaluated in patients with post-infarcted heart in clinical trials. Cardiac Arrhythmias Suppressing Trial (CAST, 1989) reported an increase in the mortality of the treated groups (flecainide and encainide) against the placebo[6]. A dramatic shift from Class I agents to Class III agents was made[[4], and pure Class III drugs were considered as ideal agents (d-sotalol) in survival test with oral d-sotalol (SWORD, 1994). The d-sotalol, which is free from β-adrenergic blocking activity but can block the rapid component of delayed rectifier outward K+ currents (IKr), only failed again with a higher mortality (3.9%) than that in the placebo (2.0%)[4,5]. The lack of success in the development of antiarrhythmic agents is likely because of the lack of awareness of the mechanisms of ion channelopathy in the diseased myocardium.
Effort to gain more insight into molecular aspects of ion channelopathy in cardiac arrhythmias in both the inherited long QT syndrom (LQTS) and cardiac remodeling of diseased hearts has been ongoing[7]. Cardiac remodeling resulting from infarction or cardiomyopathy has been targeted to investigate the mechanisms underlying cardiac arrhythmias. Aberrant molecular biology and ion channelopathy are focused on the affected myocardium. The responses to varying drug treatments are compared in amelioration and exacerbation of cardiac arrhythmias.
Different patterns of sarcolemmal ion channelopathy
Single channelopathy in LQTS Gene mutations which alters the peptide sequence of the K+ and Na+ ion channels in cardiomyocytes caused LQTS. There are three kinds of LQTS[1,7,8]: (i) downregulation of the slow component of delayed rectifier outward K+ currents (IKs) by mutations in the KvLQT1 (KCNQ1), and HMINK (KCNE1) gene; (ii) downregulation of the IKr channels by mutations in the HERG (KCNH1) gene; (iii) upregulation of the sodium currents (INa) by mutations in the SCN5A gene. Ion channelopathy induced by mutations only involves an individual channel[9,10]. Two mutations in the HERG gene were found. An early appearance of STOP codon located at the nucleotide binding domain left one dysfunctional channel responsible for LQTS, but another in the minor region of the peptide chain as a result of polymorphism did not cause the change of QT interval[10].
Multi-channelopathy in cardiac remodeling In the hypertrophied ventricle following myocardial infarction or cardiomyopathy, the multiple ion channels involving the Na+, K+ and Ca2+ currents in the lipid membrane were downregulated in function or mRNA abundance[11–13], rather than being limited to a single one[14 ]. In the pulmonary hypertension the downregulation of the genes encoding the Ito channels (Kv 4.2 and Kv 4.3) was found in the hypertrophied right ventricle, but not the left ventricle[15]. The ion channelopathy in cardiac remodeling can be referred to as multiple and non-selective channelopathy, and is likely to be secondary to the pathological lesions upstream to the ion channels, including lesions in the affected lipid membrane and the altered transmembrane signaling system[9,10]. Ion channelopathy can be regressed when lesions upstream to the ion channels are relieved.
Upregulation of ion channels in cardiomyopathy by L-thyroxin A model of cardiomyopathy in rats was induced by repeated administration of L-thyroxin for 10 d which manifested exaggerated cardiac arrhythmias after ischemia/reperfusion[16]. The arrhythmogenesis in the model was associated with remarkable cardiac remodeling and an imbalance of transmembrane distribution of cation, shown by an augmented activity of Na+/K+ ATPase and Ca2+ ATPase in the sarcolemma and mitochondria[17,18].
The channels of slow component of delayed rectifier outward K+ currents (IKs) and the rapid component of delayed rectifier outward K+ currents (IKr) were upregulated accompanied with myocardium remodeling induced by L-thyroxin[19] which would cause a shortened action potential duration (APD). However, the ICa,L was exacerbated in isolated myocytes in this model which had the potential to prolong APD[20]. The length of APD, in general, is not dependent on ions movement in a single channel, but is modulated by the balance of total currents in the repolarization of the membrane[9]. A simple mathematic equation denotes the balance between the influx and efflux of ions across the membrane: APD=(ICa,L·INa)/(IKr· IKs). The final impact on the APD is dependent on the net influence summarized by the individual ion current. Varied repolarization, which contributes to cardiac arrhythmias in a diseased heart, shows retardation and dispersion of APD. The abnormality of repolarization can be measured by the length of either APD or the QT interval in ECG traces. Dispersion of APD between the right and left ventricle in association with arrhythmogenesis was developed in a hypertrophied canine heart by chronic destruction of the atrioventricular node[21]. More dispersed ion currents in repolarization create a higher risk of developing life-threatening arrhythmias. Arrhythmic models of the canine diseased heart and rat cardiomyopathy by L-thyroxin share important properties of dispersed repolarization and disturbed multiple ion channels[21,22].
The normal function of some ion channels is modulated by phosphorylation at the nucleotide binding domains of the channel peptide, mediated by cAMP and PKA[3,22]. Hyperphosphorylation under the pathological condition serves as an important etiological factor to develop severe cardiac arrhythmias in diseased status. The upregulation of IKs, IKr, and ICa,L in cardiac remodeling by chronic administration of L-thyroxin was likely to cause overphosphorylation, so arrhythmias in this model were effectively suppressed by propranolol[16].
Ion channelopathy in sarcoplasmic reticulum
RyR2 and SERCA2a in the sarcoplasmic reticulum (SR) A rise of free calcium in the cytosol is initiated by an influx of Ca2+ through ICa,L and is amplified up to 10-fold by calcium release from the RyR2/calcium release channels, which is mediated by the calcium-induced calcium release (CICR) that promotes a boost of free calcium in the systole. A rapid conversion into the diastolic phase is performed by SERCA2a/calcium-uptaking channels. The calcium-release channels (RyR2) and the uptake channels (SERCA2a) of the SR are modulated separately by two proteins, calstabin 2 protein (FKBP12.6, the stabilizing FK 506 binding protein) and phospholamban (PLB), which are under the control of the phosphorylation of PKA[23]. The association of FKBP12.6 to the macromolecule RyR2 is essential in stabilizing the Ca2+ releasing function by tightly closing the channel against the low level of Ca2+ in the diastolic period. The movement of the free calcium from cytosol into the SR by SERCA2a is slow as a result of suppression by PLB at the resting state. The efficacy of SERCA2a increases significantly when the PLB is phosphorylated. This is caused by the removal of the suppression through PLB phosphorylation. An abnormal boost of the free calcium in the diastolic phase is provided by the derangement of either RyR2-FKBP12.6 or PLB-SERCA2a systems that play active roles in exhibiting cardiac arrhythmias during stress-related conditions[3,24,25].
Inherited arrhythmogenetic disorders from mutations of RyR2 gene There are two inherited diseases that manifest cardiac arrhythmias stemming from RyR2 gene mutations. Arrhythmigenic right ventricular dysplasia/cardiomyopathy (ARVD/C) shows distinguished morphological changes in the right ventricle where myocytes are replaced by fibrosis and adipose tissue[26,27]. Catecholaminergic polymorphic ventricular tachyarrhythmias (CPVT)[28] is another inherited disease that puts patients at risk of developing arrhythmias during physical exercise, but not in a resting state[29]. More catecholamines are released in the process of physical exercise to initiate early after depolarization (EAD) and delayed after depolarization (DAD) in these patients through a mechanism of the triggering activity, as a consequence malignant ventricular arrhythmias are more likely to occur. Mutations in the RyR2 gene attenuated the affinity of RyR2 to FKBP12.6 which resulted in tachyarrhythmias.
Hyperphosphorylation of ion channels Both sarcolemmal ion channels and calcium-release and uptake channels in the sarcoplasmic reticulum can be hyperphosphorylated.
Hyperphosphorylation of ion channels in lipid membrane There is a nucleotide binding domain in the sequence of the channel peptide of IKr, IKr, and ICa,L. Hyperphosphorylation of the channel peptide promotes an upregulation that is likely to provide a molecular basis for the varying function of the ion channels in a diseased heart. The up-regulated IKr and IKs in L-thyroxin-induced cardiomyopathy[30] contributed to the retardation of APD. In contrast, an unregulated ICa,L[31], another consequence of hyperphosphorylation, offered the potential to shorten the APD by facilitating repolarization. The co-existence of two factors to prolong or shorten APD is responsible for the dispersion of repolarization in cardiac remodeling under the control of PKA phosphorylation. The existence of hyperphosphorylation has been evidenced by a significant increase in the PKA mRNA in the injured myocardium induced by L-thyroxin.
The post-infarcted rat heart with a hypertrophied ventricle presents a mild enhancement in cardiac arrhythmias that occur in the ischemia/reperfusion procedure using Langendorff apparatus. After a treatment with isoproterenol for 5 d, the severity of cardiac arrhythmias and cardiac remodeling are greatly exacerbated, which can be attributed to PKA-hyperphosphorylation. The impact of cardiac hypertrophy on the ICa,L is uncertain, however, the intensity of the ICa,L in the infarcted and isoproterenol treated rat model is augmented significantly in the left ventricle, but not in the right ventricle. A significant dispersion of the ICa.L current by hyperphosphorylation has been found to be responsible for the exaggerated arrhythmias.
Hyperphosphorylation of RyR2-FKBP12.6 macromolecule complex Hyperphosphorylation of the RyR2 macromolecule complex that dissociates the FKBP12.6 from its binding site on the RyR2 can be developed by an overactivation of the β-adrenergic receptors[23]. This is presented in a failing heart with relevant cardiac arrhythmias[24,25]. Hyperphos-phorylation converts the normal function of RyR2 into an oversensitive state towards the low diastolic low calcium levels. A substantial leakage of Ca2+ at the diastole, which is the main result of dissociation of FKBP12.6, contributes to a partial depolarization process by a boost in Ca2+ ion in repolarization. The delayed process of repolarization causes a prolonged APD that promotes EAD and DAD, and eventually induces cardiac tachyarrhythmias.
Hyperphosphorylation of PLB-SERCA2a system A maladaptive response to PLB by hyperphosphorylation[3,32] causes a depressed SERCA2a capacity, after which elevated Ca2+ levels at the diastole are initiated. Hyperphosphoryla-tion of PLB could negatively modulate the maximal capacity of SERCA2a by means of transgenic procedure, possibly involving other phosphorylated proteins[3]. The mechanism of hyperphosphorylation might participate in arrhythmo-genesis induced by L-thyroxin where the mRNA of SERCA is altered significantly.
The post-infarcted rat heart shows a mild-depressed SERCA2a mRNA and less severe cardiac arrhythmias, possibly reflecting hyperphosphorylation to a lesser extent[33]. After a 5-d treatment with isoproterenol, the infarcted heart was converted into a significantly augmented arrhythmia accompanied with more depressed mRNA of SERCA2a which was mediated by PKA-hyperphosphorylation.
Dispersion of the calcium-handling system by over-phosphorylation We demonstrated that the significant dispersion of depressed mRNA abundance of RyR2 and SERCA and upregulated mRNA abundance of sodium calcium exchanger (NCX) in infarcted heart was associated with an increase in arrhythmic score after an isoproterenol treatment. The mRNA abundance of RyR2 and SERCA2 were decreased by 83.6% and 77.6% in left ventricle and by 36.7% and 38.1% in right ventricle, respectively. The mRNA abundance of NCX1 was increased by 63.8% in left ventricle and 40.4% in right ventricle. The difference between left and right ventricle was significant. This implies that the free calcium levels in the left ventricle could be relatively higher than in the right ventricle under hyperphosphorylation. An elevated diastolic calcium level is likely to be the result of an alternative route, which could be sourced by an increase in NCX under hyperphosphorylation. An up-regulated NCX mRNA in the left ventricle possibly indicates more calcium than that in the right ventricle[33]. A dispersion of the calcium-handling system under the control of PKA-overphosphorylation in an infarcted heart can be the basis for initiating cardiac arrhythmias under stress. The onset of severe cardiac arrhythmias based on not only the dispersion of the ion channels in the lipid membrane, but also the release, uptake, and exchange of the calcium-handling system.
The maladjustment in RyR2-FKBP12.6 and SERCA2a-PLB by hyperphosphorylation happened in various conditions (eg, in failing hearts and over-stimulation of the β-adrenergic receptors by activated sympathetic impulses). Sudden cardiac death results from the over-activation of PKA[34]. The aberrant RyR2-FKBP12.6 and SERCA2a-PLB are important to develop malignant arrhythmias in an affected heart and serve as new targets for drug intervention to suppress cardiac tachyarrhythmias. The severely exaggerated cardiac arrhythmias provoked by an ischemia/reperfusion episode in the hypertrophied ventricle are produced either by repeated administration of L-thyroxine or isoproterenol in chronic infarcted rat heart. These could be related to the dispersion of deranged activity of the intracellular calcium-handling system as a consequence of hyperphosphorylation of the SR.
Direct and indirect effects of drugs on ion channels
Direct effect on the ion channels The effects of antiarrhythmic agents on ion channels could be recognized as either, a direct action on ion channels or an indirect effect, with which an improvement can be achieved on the altered ion channels by relieving the upstream lesions. Using the regular micro-electrode and patch-clamp techniques we observed that CPU 86017 blocked sodium, potassium, and calcium channels. A biphasic effect on the APD is suggested to be the result of a multi-channel blockade[35]. So CPU 86017 could modulate APD in two directions, either prolonging the already shortened APD or shortening the already lengthened APD caused by lesions in a diseased myocardium. Therefore, reducing the dispersion of APD in a diseased heart to prevent cardiac arrhythmias is hopeful[36]. Prolonging APD by anti-arrhythmic agents is not beneficial for inhibiting ventricular tachyarrhythmias. In contrast, it always implies a potential of an adverse reaction by producing an increased incidence of Torsades de pointes (TdP)[37], a dangerous ventricular tachycardia with a greater tendency to ventricular fibrillation.
Most electrophysiological data of anti-arrhythmic agents on individual ion channels are collected from freshly isolated myocytes. However, cardiac arrhythmias appear frequently in diseased and remodeled rather than normal hearts, and the molecular biological properties of the myocardium are markedly different in many aspects between the normal and diseased hearts. Dispersed repolarization stems from disturbed channels in the diseased myocardium only. It is worthy of investigating drug effects on altered ion currents in isolated myocytes of diseased hearts.
Indirect effects on ion channelopathy Cardiac arrhythmias could be suppressed by chronic drug therapy for restoring the altered channels. The restoration of up-regulation of the IKr, IKs and ICa.L current of isolated myocytes is confirmed by chronic medication of the propranolol, bepridil, and CPU 86017 in L-thyroxin-induced cardiomyopathy[30,31].
The ion channels in a chronically infarcted heart is mostly down-regulated. Therefore, it is confusing to face the fact that an anti-arrhythmic agent could stop tachyarrhythmias by overlapping its suppressive effect on a depressed channel in a remodeled myocardium. There should be some up-regulated ion channels in diseased hearts. The up-regulated channels, which are denoted clearly in the cardiomyopathy induced by L-thyroxin, are linked with the occurrence of cardiac arrhythmias and serve as the target for anti-arrhythmic therapy. The action of drugs on the ion channels can be described as direct and chronic rather than a direct effect alone (Figure 1). The ability of drugs to restore the diseased channels either mediated by an indirect action on the upstream lesion to the ion channels via chronic medication or an acute suppression on an individual current contributes to the correction of disordered channels and prevention from the sudden episode of cardiac death resultant from deterioration of ventricular fibrillation on stress-related events.
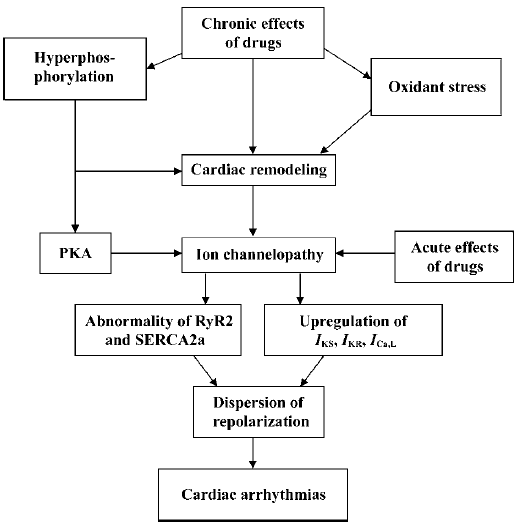
Phases of ion channelopathy
Two phases of ion channelopathy The pathological progression of ion channels in the myocardium either in the sarcolemmal membrane or in the SR could be subdivided into two different stages: the basal phase, in which no presentation of arrhythmias can be found; and the activated phase, in which arrhythmias appear when ion channelopathy is deteriorated.
Basal channelopathy with disturbed ion channels, which indicate an abnormal repolarization in the myocardium, manifests no arrhythmia. This is because of a reservoir of repolarization. The normal period of repolarization is less than 460 ms and an APD/QTc is referred as prolonged when beyond 460 ms. Malignant arrhythmias are most likely to occur when an APD/QTc is longer than 520 ms. A prolongation of APD/QTc in the range of 460 ms to 520 ms is considered as a repolarization reservoir at risk of developing TDP, however, there is no arrhythmia at the resting state. It is common that patients who either inherit mutation in genes of the LQTS, the ARVD/C, and CPVT, or possess predominant cardiac remodeling, exhibit no arrhythmia at the resting state, suggesting that prolonged APD/QTc is still within the repolarization reservoir. However, if an APD/QTc is prolonged over 520 ms, the incidence of malignant arrhythmias is very high and arrhythmias are more likely to occur sooner.
Ion channelopathy can be deteriorated. The occurrence of ventricular tachyarrhythmias needs an activation of the channelopathy accompanied with an over-activation of the β-adrenergic receptors induced by heavy physical or mental stress. The process involves a burst of oxidative stress and an imbalance of the oxygen supply and demand in the diseased myocardium. Hyperphosphorylation of RyR2 and PLB is involved in the exaggeration of the channelopathy by dissociating the FKBP12.6 and depressing the activity of SERCA2a, respectively. The process is not merely induced by the activation of β-adrenergic receptors, and the oxidative stress and activation of the ET-1 system and rennin-angiotension system are likely to mediate the activation pathological process.
Many kinds of drugs, such as ACEI, AT1 receptor blockers, endothelin receptor antagonists, the β-blockers, and anti-oxidative agents have antiarrhythmic effects by diminishing upstream lesions to relieve related channelopathy (ie, the cardiac remodeling and the abnormality of the transmembrane signaling system). Endothelins are biosynthesized and released in a substantial amount from the diseased myocardium and exert harmful effects on myocardium by stimulating cell proliferation, inducing apoptosis, and activating the production of free radicals. An excess of ET-1 promotes oxidative stress which is mediated by an increase in iNOS activity to form peroxynitrite ONOO¯· . Peroxynitrite is cytotoxic to myocardium and is likely to take an active part in the progression of ion channelopathy and cardiac arrhythmias. The endothelin ETA/ETB receptor antagonists reduced cardiac remodeling and cardiac failure by blocking the overactivation of the ET system[37–39]. It is interesting to find that an endothelin receptor antagonist, Dajisentan (CPU 0213), is effective in suppressing cardiac arrhythmias after ischemia/reperfusion in L-thyroxin-induced cardiomyopathy, but has no effect on ischemia/reperfusion-induced arrhythmias in a normal rat heart.
Class III anti-arrhythmic agents
In controlling ventricular tachyarrhythmias the Class I agents (INa blocking agents) are not recommended to patients with a diseased ventricle because of their strong potency to induce arrhythmias by suppressing the propagation of impulse[40]. It is still questionable whether pure Class III antiarrhythmic agents would suppress ventricular tachyarrhythmias effectively and safely in a diseased heart. To date, the common opinion is that it is favorable to use the complex Class III agents, such as a low dose of amiodarone, rather than the pure Class III agents in treating ventricular tachyarrhythmias (Figure 2).
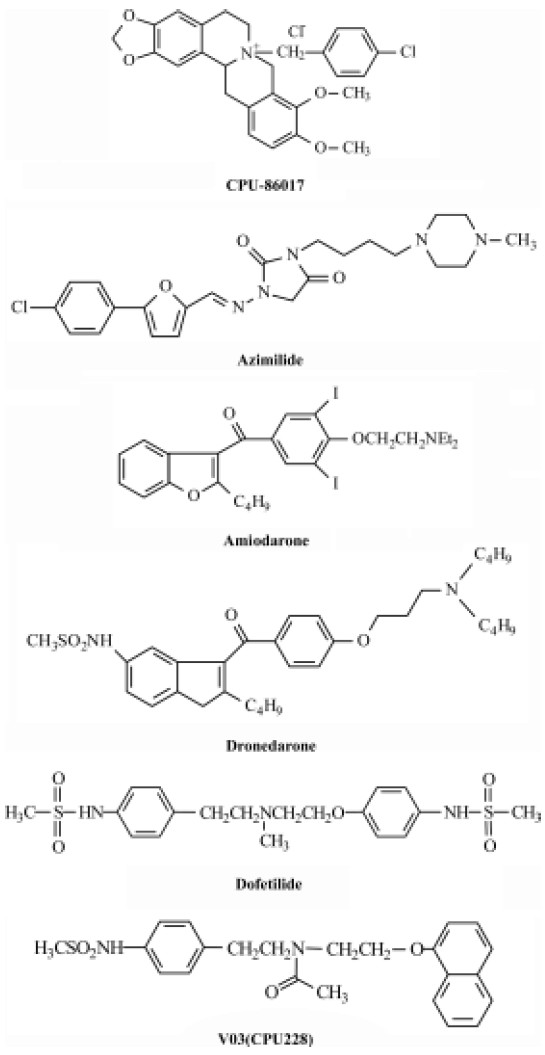
Pure Class III agents Dofetilide and ibutilide belong to pure Class III agents that prolong APD remarkably by inhibiting the IKr currents only[41,42]. The two agents are favorable in the treatment of atrial fibrillation and flutter, but not in the control of ventricular tachyarrhythmias. Physicians should take care of monitoring the QTc in treatment of atrial flutter/fibrillation because of the high incidence of Torsades de Pointes when using the two agents. The outcome of clinical trials of dofetilide on mortality in the treatment of ventricular arrhythmias is neutral[40,42].
Complex Class III agents Complex Class III anti-arrhythmic agents block not only IKr currents but also other channels and receptors.
Complex Class III anti-arrhythmic agents block IKs. These include amiodarone, dronedarone, azimilide, CPU 86017, and so on. Blocking IKs is important to shorten the APD and control cardiac arrhythmias by reducing the interference of β-receptor stimulation that causes an exacerbated IKs.
Complex Class III anti-arrhythmic agents block ICa,L. These include amiodarone[43], dronedarone[44], azimilide[45], and CPU 86017[46]. An upregulation of the ICa,L channels is associated with an enhancement of arrhythmogenesis in L-thyroxin-induced cardiac remodeling[20] and in an infarcted heart after isoproterenol treatment[31]. Blocking ICa,L is beneficial to control arrhythmia by relieving calcium overload and restoring a disordered calcium-handling system in the diseased myocytes. In addition, an inhibition of the calcium current helps to prevent cardiac arrhythmias by restoring the imbalance of the altered ion currents during repolarization in diseased hearts.
Dofetilide has the potential to develop TdP by its tremendous effect in prolonging APD. A combination with an INa blocking agent was successful in attenuating the incidence of TdP induced by a pure Class III agent[47]. According to the mathematic equation to calculate the length of APD mentioned previously, we hypothesize that shortening in APD by blocking ICa,L could reduce the incidence of TdP. Therefore, CPU 228 was created by modification on the moiety of dofetilide. Treatment with CPU 228 significantly decrease the incidence of TdP in the rabbit model[41]. CPU 228 can block both ICa,L and IKr, so it is being converted from a pure Class III antiarrhythmic agent, dofetilide, into a complex Class III agent that has more potency to control arrhythmias in an animal model.
Conclusion
The concepts and targets for the development of antiarrhythmic drugs are evolving[48]. It is hopeful that more safe and effective agents will be obtained from continuous efforts based on regressing and relieving the basal ion channelopathy and preventing the deterioration of channelopathy in the view of biomolecular mechanisms. Propranolol and verapamil inhibit mRNA expression of RyR2 and SERCA in L-thyroxin-induced rat ventricular hypertrophyl[49]. β-Adrenergic stimulation increases current amplitude and activation of ICa-L[31] and IKs[50], causing dispersion of repolarization. More research at the molecular level will be needed on ion channelopathy. The safe and effective agents in controlling life-threatening arrhythmias can be obtained by relieving upstream lesions to ion channels, mainly through anti-hyperphosphorylation.
References
- Roden DM, Balser JR, George AL Jr, Anderson ME. Cardiac ion channels. Ann Rev Physiol 2002;64:431-75.
- Marban E. Cardiac channelopathies. Nature 2002;415:213-8.
- Olson EN. A decade of discoveries in cardiac biology. Nat Med 2004;10:467-74.
- Colastsky TJ. Antiarrhythmic drugs: where are we going? Pharm News 1995;2:17-23.
- Guerra PG, Talajic M, Roy D, Dubuc M, Thibault B, Nattel S. Is there a future for antiarrhythmic drug therapy? Drugs 1998;56:767-81.
- The Cardiac Arrhythmia Suppression Trial Investigations. Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmias suppression after myocardial infarction. N Engl J Med 1989;321:406-12.
- Dumaine R, Antzelevitch C. Molecular mechanisms underlying the long QT syndrome. Curr Opin Cardiol 2002;17:36-42.
- Roden DM, Lazzara R, Rosen M, Schwartz PJ, Towbin J, Vincent GM, et al. Multiple mechanisms in the long-QT syndrome current knowledge, gaps, and future directions. Circulation 1996;94:1996-2012.
- Dai DZ. Vulnerable substrate and multiple ion channel disorder in a disease heart will be new targets for antiarrhythmic therapy. Acta Pharmacol Sin 2000;21:289-95.
- Dai DZ. Disorder ion channels and antiarrhythmic agents. Pharm News 2001;8:1-6.
- abauer M, Kaab S. Potassium channel down-regulation in heart failure. Cardiovasc Res 1998;37:324-34.
- Qin DY, Zhang ZH, Caref EB, Boutjdir M, Jain P, Ei-Sherif N. Cellular and ionic basis of arrhythmias in post-infarction remodeled ventricular myocardium. Circ Res 1996;79:461-73.
- Dai DZ. The ion channelopathy and reconsideration of antiarrhythmic drugs. In: Su DF, Miao ZY, Wang YM, editors. Advances in Pharmacology. Beijing: People’s Medical Publishing House; 2002. p 202−11.
- Dai DZ, Hu HQ, Yang P, Ma YP, Cao W, Wang H. The proarrhythmic activity and the ion channelopathy in remodeled myocardium. Chin J Pathophysiol 2000;16:945-8.
- Zhang TT, Cui B, Dai DZ. Down-regulation of Kv4.2 and Kv4.3 channel gene expression in right ventricular hypertrophy induced by monocrotaline in rats. Acta Pharmacol Sin 2004;25:226-30.
- Yu F, Dai DZ, An LF, Guo XF. Heart hypertrophy induced by levothyroxin aggravates ischemic lesions and reperfusion arrhythmias in rats. Acta Pharmacol Sin 1997;18:71-4.
- Chen DD, Dai DZ, Wang CX. Effects of verapamil and captopril on cardiac hypertrophy and elevated left ventricular sarcolemmal Na+,K+-ATPase activity induced by L-thyroxin in rats. Chin J Pharmacol Toxicol 1995;9:228-30.
- Chen DD, Dai DZ, Lu J, Zhang XK. Propranolol and bepridil attenuating levothyroxin-induced rat cardiac hypertrophy and mitochondrial Ca2+-Mg2+-ATPase activity elevation. Acta Pharmacol Sin 1996;17:516-8.
- Ma YP, Hao XM, Zhang GQ, Zhou PA, Wu CH, Dai DZ. Blocking effect of CPU 86017 on slowly activated rectifier potassium current in single ventricular myocytes of guinea pigs. J Chin Pharm Univ 2000;31:121-5.
- Dai DZ, Hu HJ, Yang DM, Hao XM, Zhang GQ, Zhou PA, et al. Chronic levothyroxin treatment in associated with ion channel abnormalities in cardiac and neuronal cells. Clin Exp Pharmacol Physiol 1999;26:819-21.
- Volder PGA, Sipido KR, Vos MA. Downregulation of delayed rectifier K+ current in dogs with chronic complete atrioventricular block and acquired Torsades de Pointes. Circulation 1999;100:2455-61.
- Marx SO, Reiken S, Hisamatsu Y, Jayamana T, Burkhoff D, Rosemblit N, et al. PKA phosphorylation dissociated FKBP12.6 from the calcium release channel (ryanodine receptor) defective regulation in failing hearts. Cell 2000;101:365-76.
- Wehrens XH, Lehnart SE, Huang F, Vest JA, Reiken SR, Mohler PJ, et al. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell 2003;113:829-40.
- Scoote M, Willians AJ. The cardiac ryanodine receptor (calcium release channel): emerging role in heart failure and arrhythmia pathogenesis. Cardiovasc Res 2002;56:359-72.
- Paul M, Schulze-Bahr E, Breithardt G, Wichter T. Genetics of arrhythmogenic right ventricular cardiomyopathy−status quo and future perspectives. Z Kardiol 2003;92:3628-49.
- Ahmad F. The molecular genetics of arrhythmogenic right ventricular dysplasia-cardiomyopathy. Clin Invest Med 2003;26:167-78.
- Dulac Y, Acar P, Delay M. Diagnosis of adrenergic ventricular tachycardia in two homozygotic twins. Arch Mal Coeur Vaiss 2003;96:521-3.
- Lahat H, Pras E, Eldar M. RYR2 and CASQ2 mutations in patients suffering from catecholaminergic polymorphic ventricular tachycardia. Circulation 2003;107:e29.
- Dai DZ, Zhang GQ, Yang P, Ma YP. Two patterns of ion channelopathies relating to arrhythmias and direct and indirect blockade of ion channels by antiarrhythmic agents. Drug Dev Res 2003;58:42-50.
- Wang HL, Li SB, Dai DZ. Change of L-type calcium current in single guinea pig hypertrophic ventricular myocytes induced by levothyroxin. J Chin Pharm Univ 2000;31:130-4.
- Hort BD, Tramuta DA, Kadambi VJ, Dash R, Ball N, Kranias EG, et al. Influence of transgenic overexpression of phospholamban on post-extrasystolic potentiation. J Mol Cell Cardiol 1999;31:2007-15.
- Wang HL, Dai DZ, Gao F, Zhang YP, Lu F. Dispersion of ventricular mRNA of RyR2, and SERCA associated with arrhythmo-genesis in rats. Acta Pharmacol Sin 2004;25:738-43.
- Antos CL, Frey N, Marx SO, Reiken S, Gaburjakova M, Richardson JA, et al. Dilated cardiomyopathy and sudden death resulting from constitutive activation of protein kinase A. Circ Res 2001;89:997-1004.
- Dai DZ, Yu F, Li HT, Tang YQ, An LF, Huang WL, et al. Blockade on sodium, potassium, and calcium channels by a new antiarrhythmic agent CPU 86017. Drug Dev Res 1996;39:138-46.
- Lu HR, Yu F, Dai DZ, Remeysen P, De Clerck F. Reduction in QT dispersion and ventricular arrhythmias by ischaemic preconditioning in anaesthetized, normotensive and spontaneously hypertensive rats. Fundam Clin Pharmacol 1999;13:445-54.
- Guillermo TA, Young JB, Durand JB, Bozkurt B, Mann DL, Kobrin I, et al. Hemodynamic effects of Tezosentan, an intravenous dual endothelin receptor antagonist, in patients with class III to IV congestive heart failure. Circulation 2001;103:973-80.
- Miyauchi T, Goto K. Heart failure and endothelin receptor antagonists. Trend Pharmacol Sci 1999;20:210-7.
- Dai DZ, Huang M, Ji M, Liu LG. A novel endothelin receptor antagonist CPU 0213 and therapeutic effect on acute heart failure in rats. J China Pharm Univ 2005;36. in press.
- Van Gelder IC, Brugemann J, Crijns HJ. Current treatment recommendations in antiarrhythmic therapy. Drugs 1998;55:331-46.
- Howard PA. Ibutilide: an antiarrhythmic agent for the treatment of atrial fibrillation or flutter. Ann Pharmacother 1999;33:38-47.
- Brendorp B, Elming H, Jun L, Kober L, Torp-Pedersen C. Diamond Study Group. The prognostic value of QTc interval and QT dispersion following myocardial infarction in patients treated with or without dofetilide. Clin Cardiol 2003;26:219-25.
- Yoshida H, Sugiyama A, Satoh Y, Ishida Y, Yoneyama M, Kugiyama K, et al. Comparison of the in vivo electrophysiological and proarrhyhmic effects of amiodarone with those of a selective class III drug, sematilide, using a canine chronic atrioventricular block model. Circ J 2002;66:75-62.
- Doggrell SA, Hancox JC. Dronedarone: an amiodarone analogue. Expert Opin Investig Drugs 2004;13:415-26.
- Fermini B, Jurkiewicz NK, Jow B, Guinosso PJ, Baskin EP, Lynch JJ, et al. Use-dependent effects of the class III antiarrhythmic agent NE-10064 (Azimilide) on cardiac repolarization: block of delayed rectifier potassium and L-type calcium currents. J Cardiovasc Pharmacol 1995;26:259-71.
- Dai DZ. The anti-arrhythmic activity of protoberberines in relation to blockade of ion channels. Ion Channel Modulators 1997;2:383-90.
- Hallman K, Carlsson L. Prevention of class III– induced pro-arrhythmias by flecainide in an animal model of the acquired long QT syndrome. Pharmacol Toxicol 1995;77:250-4.
- Nemec J, Shen WK. Antiarrhythmic drugs: new agents and evolving concepts. Expert Opin Investig Drugs 2003;12:435-53.
- Wu XD, Dai DZ, Zhang GQ, Gao F. Propranolol and verapamil inhibit mRNA expression of RyR2 and SERCA in L-thyroxin-induced rat ventricular hypertrophy. Acta Pharmacol Sin 2004;25:347-51.
- Han W, Wang Z, Nattel S. Slow delayed rectifier current and repolarization in canine cardiac Purkinje cells. Am J Physiol Heart Circ Physiol 2001;280:H1075-80.