
Nitric oxide modulates hypoxic pulmonary smooth muscle cell proliferation and apoptosis by regulating carbon monoxide pathway1
Introduction
The proliferation and apoptosis of vascular smooth muscle cells (VSMC) maintain cellular homeostasis of the vessel wall, and the imbalance between them constitutes the cytological basis of vascular structural remodeling in a variety of vascular diseases including hypoxic pulmonary hypertension. In response to chronic hypoxia, promoted proliferation and inhibited apoptosis of smooth muscle cells in pulmonary arteries occur[1–3], which eventually leads to vascular remodeling. However, the underlying mechanism of pulmonary artery smooth muscle cells (PASMC) response to hypoxia and its regulations remains to be clarified.
Studies have demonstrated that endothelium-derived nitric oxide (NO), which is synthesized from L-arginine by endothelial nitric oxide synthase (eNOS) in vascular endothelial cells, attenuated pulmonary vascular structural remodeling by inhibiting proliferation and promoting apoptosis of rat PASMC[4–7]. Heme oxygenase (HO)-1 derived carbon monoxide (CO), which is catalyzed from heme mainly by VSMC, has also been known to have antiproliferative and pro-apoptotic effects on hypoxic PASMC[8–10].
Recent evidence suggested that NO was a potent inducer of HO-1 gene expression in VSMC, resulting in an increase in production of CO, which in turn inhibited cellular proliferation[11–16]. Since NO has been shown to exert antiproliferative and pro-apoptotic effects on PASMC, it was reasonable to propose that the HO-1/CO pathway might be involved in the regulation of hypoxic pulmonary vascular structural remodeling by NO. Whether HO-1 expression contributes to the antiproliferative and/or pro-apoptotic effects mediated by NO remains to be investigated. We hypothesized that NO and CO interact to regulate the proliferation and apoptosis of hypoxic PASMC. The present study was designed on the cultured rat PASMC to observe the effects of hypoxia on CO and NO generation, and the effects of NO and CO on cellular proliferation and apoptosis, and to elucidate the role of CO in NO-regulated PASMC proliferation and apoptosis induced by hypoxia.
Materials and methods
Materials Male Wistar rats, weighing 150–180 g, were obtained from Peking University Animal Center (Beijing, China). Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), trypsin and Trizol were from Gibco-BRL Co (Gaithersburg, MD, USA). Zinc protoporphyrin-IX (ZnPP-IX), sodium nitroprusside (SNP) and calf hemoglobin (Hb) were obtained from Sigma Chemical Co (St Louis, MO, USA). ZnPP-IX was dissolved in 50 mmol/L fresh sodium carbonate, and SNP was dissolved by fresh sterile water. Proliferating cell nuclear antigen (PCNA) monoclonal antibody and caspase-3 polyclonal antibody were from Zhongshan Bio-tech Co (Beijing, China). Moloney murine leukemia virus (M-MLV) and Taq DNA polymerase were purchased from Promega Co (Madison, WI, USA).
PASMC culture The rats were anesthetized with 0.25% pentobarbital sodium (40 mg/kg, intraperitoneal injection). The main pulmonary artery and its branches were stripped of adventitia and the PASMC was removed to culture media sterilely. All cells were grown in DMEM supplemented with 10% heat-inactivated FBS, 100 U/mL penicillin, and 100 U/mL streptomycin. The cells were processed twice a week by harvesting with trypsin and seeded (1:3 ratio) into 75 cm2 flasks. For the experiments, subcultured cells were seeded into multi-well plates or 100 mm culture dishes and used between passages 5 and 10. The cells were identified by VSMC-α-actin immunological criteria. All cultures were maintained at 37 °C in a humidified atmosphere of 5% CO2–95% air. When the cells reached 80% confluence (3 or 4 d), the culture media were replaced with serum-free DMEM containing FBS (0.1%, w/v) for 24 h and then exposed to the various treatment regimens (either solvent or drugs) prior to hypoxic exposure. The hypoxic gas mixture (95% N2, 5% CO2) was pre-analyzed and infused into airtight incubators with in-flow and out-flow valves (Billups-Rothenberg, Del Mar, CA, USA) at a flow rate of 3 L/min for 15 min to attain a PO2 of 18–20 mmHg. Unless otherwise stated, each group shown in the text was composed of 8 rats.
Fluorescent real-time quantitative RT-PCR RT-PCR assay was performed with 106 cells. Total RNA was extracted from cultured PASMC using Trizol reagent as described in the handbook (Gibco-BRL, USA), with direct lysis of cells in Trizol lysis buffer followed by chloroform extraction. Ethidium bromide staining of the 1.5% gel was used to confirm RNA integrity. Ten microliters of RNA solution were used for RT using oligo dT and M-MLV reverse transcriptase. First strand cDNA synthesis was obtained after 1 h at 42 °C. DNA synthesis was performed using an automatic thermo-cycle (GeneAmp 5700, USA). Samples were subjected to 30 cycles of fluorescent RT-PCR. The primer was designed to amplify the 176 bp sequence. The oligonucleotide primer sequences (HO-1 S: 5'-TCT ATC GTG CTC GCA TGA AC-3' and HO-1 A: 5'-CCT CTG GCG AAG AAA CTC TG-3') were designed according to Gene Bank [17]. The amplified DNA was electrophoresed on 1.5% agarose gel in tris-acetic acid-EDTA buffer and visualized by staining with ethidium bromide. A control sample without cDNA was also included in this assay to check for possible contamination. All results were analyzed automatically by GeneAmp 5700 software (PE Applied Biosystems, Foster, CA, USA).
Immunocytochemistry The rat PASMC, inoculated on glass slides, was incubated in DMEM and exposed to various treatment regimens for 12 or 24 h. The cells were washed 3 times in phosphate buffered solution (PBS), fixed in cool acetone at room temperature for 20 min, and washed in PBS 3 times again. After that, the cells were incubated with goat serum for 30 min at room temperature. The reaction was terminated with 0.1% Triton X-100 for 5 min. The cells were then incubated overnight at 4 °C with either a monoclonal antibody to PCNA (1:150 diluted) or polyclonal antibody caspase-3 (1:150 diluted). The sections were washed 3 times with PBS and incubated for 1 h with secondary antibody (biotinylated goat anti-mouse IgG working solution) at room temperature. They were rinsed 3 times with PBS. The signal was detected with peroxidase-conjugated avidin-biotin for 30 min. PBS was then used to stop the reaction. For negative controls, sections were processed as described, except that the primary incubation was performed with nonimmune rabbit serum instead of primary antibodies. The sections were then developed for color in 3,3'-diaminobenzidine for 5–10 min and mounted onto slides for microscopic observa-tion. The presence of brown granules in PASMC was defined as a positive signal. Five high power fields were selected to calculate the proportion of positive cells, according to the following formula: the percentage of positive cells (%) =the number of positive cells/the number of total cells 100%.
Measurement of nitrite The generation of NO was determined by measuring the release of nitrite (NO2–); the stable oxidation product of NO sample aliquots (0.4 mL) was mixed with an equal volume of Greiss reagent (1% sulfanilamide and 1% naphthylenediamine dihydrochloride in 2% phosphoric acid). The mixture was incubated at room temperature for 10 min and absorbance was measured at 550 nm. The concentrations of NO2– were determined with a standard curve created by using an aqueous solution of sodium nitrite. Background nitrite values corresponding to serum-free medium were subtracted from the experimental values.
Measurement of carboxyhemoglobin (HbCO) To examine the relative amount of CO being released from PASMC into the medium, Hb (50 µmol/L) was added to the cells during the last hour of incubation, and HbCO was measured by spectrophotometric method[8,18]. Difference of absorbance at 573.5 nm and 582.5 nm was read via the double-wavelength spectrophotometer. Both the wavelengths were selected according to the absorbance spectrum of HbCO and oxygenated Hb (HbO2), at which the difference of absorbance of HbCO was largest and the difference of absorbance of HbO2 was 0. Then the percentage of HbCO was calculated from a standard curve [%HbCO=(ΔOD–0.03858)/0.0498] derived by mixing different proportions of 2 Hb solutions containing 100% HbCO and 100% HbO2. We obtained these solutions by gassing Hb solutions with pure oxygen and CO, respectively. The concentration of CO was calculated according to the formula as follows: CO (µmol/L)=% HbCO× Hb (mg/L)×4000/(64 456×100×0.5).
Flow cytometry The cells were trypsinized and the cell pellet was washed with PBS. The cells were then fixed and permeated with 70% ethanol at 4 °C overnight and washed again with PBS. The pellet was resuspended in PBS containing 100 µg/mL propidium iodide and 200 µg/mL RNase A and incubated for 30 min at 37 °C. The DNA content per cell was then evaluated using a FACSortTM equipped with CELLQuestTM software (Becton Dickinson, San Jose, CA, USA). A minimum of 1×104 cells was analyzed per sample. The proportions of cells in stage G1, S and G2 were calculated in 1×104 cells examined. PASMC proliferative index (PI) and apoptotic index (AI) were calculated. The PI indicating the proliferation of PASMC was calculated using the formula: PI=[(S+G2+M)/(G0+G1+S+G2+M)]×100%. The AI detected SubG1 peaks on flow cytometry.
Statistical analysis Data were expressed as mean±SD. Statistical differences were evaluated by one-way ANOVA and then Student-Newman-Keuls test; comparisons between 2 groups involved use of the unpaired Student’s t-test. P<0.05 was considered statistically significant.
Results
Effect of hypoxia on CO release from rat PASMC The production of CO increased markedly in the presence of hypoxia at both 12 h (0.56±0.03 vs normoxia 0.40±0.02 µmol/L, P<0.01) and 24 h (0.52±0.01 vs normoxia 0.42±0.01 µmol/L, P<0.01; Table 1). When pretreated with ZnPP-IX (20 µmol/L), an inhibitor of HO, the CO production in hypoxic cells were significantly lower than that in hypoxic cells alone at both 12 h (0.30±0.02 vs 0.56±0.03 µmol/L, P<0.01) and 24 h (0.27±0.01 vs 0.52±0.01 µmol/L, P<0.01; Table 1).
Full table
Effect of hypoxia on HO-1 mRNA expression by rat PASMC Rat PASMC was exposed to hypoxia or normoxia. The levels of HO-1 mRNA in PASMC, analyzed by fluorescent real-time quantitative RT-PCR, exposed to hypoxia for 12 h demonstrated a 2-fold increase (P<0.01), compared to that of the normoxic controls. Although the level of HO-1 mRNA in the hypoxic cells declined slightly by 24 h, compared with 12 h, it remained higher than that in the normoxic controls (Figure 1).
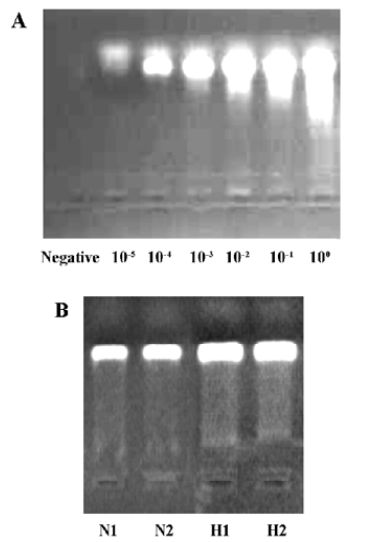
Impact of endogenous CO on the proliferation of PASMC under hypoxia Hypoxia promoted PASMC proliferation, made evident by an obvious increase in PI at 24 h (Figure 2) and PCNA immunopositive staining at both 12 h and 24 h (Table 1). When CO production by hypoxic PASMC was inhibited by pretreatment with ZnPP-IX, PI and the percentage of PCNA positive staining increased further at both 12 h and 24 h (Figure 2; Table 1).
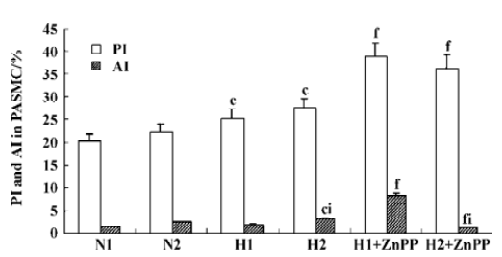
Impact of endogenous CO on the apoptosis of PASMC under hypoxia It was found that apoptosis of PASMC induced by hypoxia was more obvious at 24 h than 12 h (Figure 2; Table 1). Apoptosis of hypoxic PASMC increased further when treated with ZnPP at 12 h, compared with hypoxia alone, made evident by an increased AI. However, by 24 h, the value of AI and the expression of caspase-3 in PASMC became lower than that exposed to hypoxia alone (P<0.01; Figure 2; Table 1).
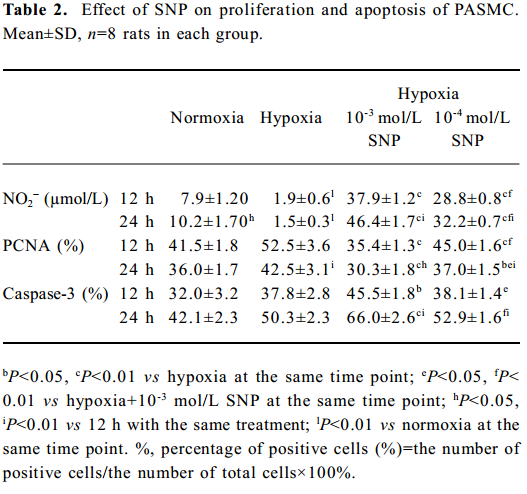
Effect of hypoxia on NO release from rat PASMC NO production from rat PASMC decreased significantly in the presence of hypoxia at both 12 h (1.9±0.6 vs normoxia 7.9±1.20 µmol/L, P<0.01) and 24 h (1.5±0.3 vs normoxia 10.2±1.70 µmol/L, P<0.01), and the low level of NO in the hypoxic environment increased markedly in the presence of SNP in a time- and concentration-dependent manner (Table 2).
Full table
Effect of NO on the growth of hypoxic PASMC Hypoxic PASMC proliferation was inhibited along with a decrease in PCNA immunostaining and PI at both 12 h and 24 h when treated with hypoxia plus SNP (Table 2; Figure 3). On the other hand, AI increased significantly in hypoxic PASMC treated with SNP, which paralleled the changes in caspase-3 immunopositive cells in number (Table 2; Figure 3).
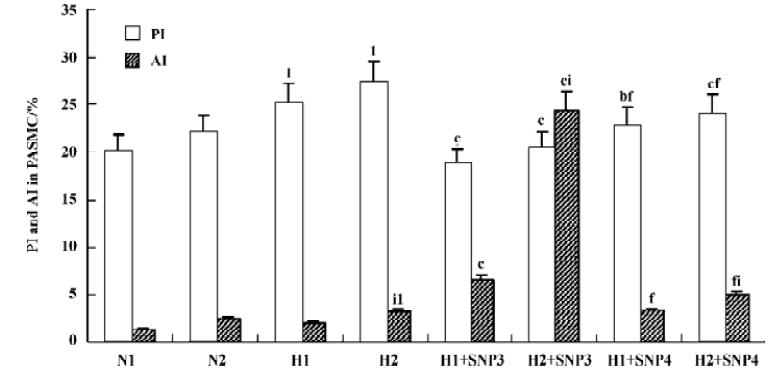
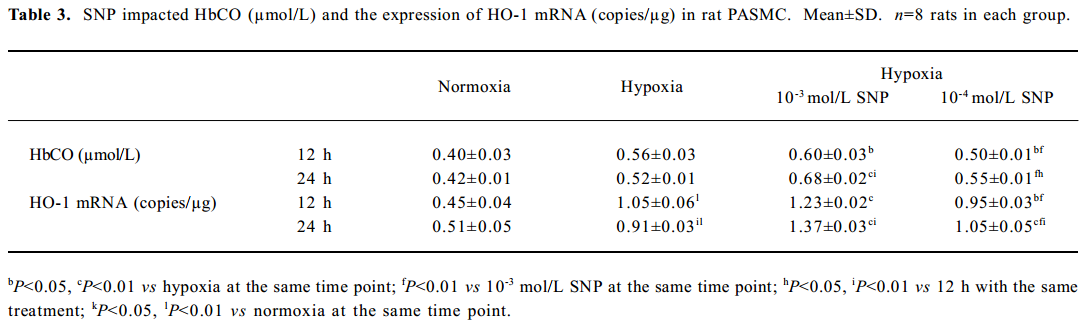
Effect of NO on CO release by rat PASMC Under both normoxic and hypoxic conditions, CO production was shown to increase after administration of SNP in both normoxic (0.55±0.01 vs 0.40±0.03 µmol/L at 12 h with 10-3 mol/L SNP, P<0.01) and hypoxic cells (0.60±0.03 vs 0.56±0.03 µmol/L at 12 h with 10-3 mol/L SNP, P<0.05). The levels of CO production increased in a time- and concentration-dependent manner (Table 3).
Full table
Effect of NO on HO-1 mRNA expression by rat PASMC under both normoxic and hypoxic conditions The levels of HO-1 mRNA after treatment with 10-3 and 10-4 mol/L SNP under normoxic and hypoxic conditions are shown in Table 3. The HO-1 mRNA level increased in a time- and concentration-dependent manner. The range of the increase in HO-1 mRNA level in the presence of 10-3 mol/L SNP was significantly greater than that in the presence of 10-4 mol/L SNP at both 12 h and 24 h. In the presence of SNP, the levels of HO-1 mRNA were higher in PASMC exposed to hypoxia for 24 h than that for 12 h.
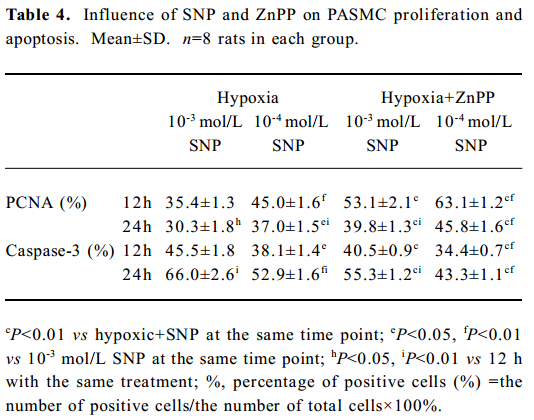
Effect of ZnPP on the regulation of PASMC PCNA and caspase-3 by SNP Compared with the expression of PCNA by hypoxic PASMC when incubated with SNP alone, the PCNA expression markedly increased (P<0.01; Table 4) when hypoxic PASMC was treated with SNP plus ZnPP. In the absence of ZnPP, PCNA expressions of hypoxic PASMC pretreated with SNP (10-3 or 10-4 mol/L) were increased at both 12 h and 24 h, respectively, compared with hypoxia alone. In the presence of ZnPP (20 µmol/L), however, they were higher than those in hypoxia plus SNP in the absence of ZnPP, respectively (Table 4).
Full table
Compared with incubation with SNP alone, caspase-3 immunostaining of hypoxic PASMC markedly decreased when it was treated with ZnPP (20 µmol/L) and SNP (P<0.01), and the index increased in a time- and concentration-dependent manner (Table 4). In the absence of ZnPP, caspase-3 expressions of hypoxic PASMC pretreated with SNP at 10-3 and 10-4 mol/L were 45.5%±1.8% and 38.1%± 1.4% at 12 h, respectively. In the presence of ZnPP, however, they were 40.5%±0.9% and 34.4%±0.7%, respectively, which were lower than those in the absence of ZnPP (all P<0.01). In the absence of ZnPP, caspase-3 expressions of hypoxic PASMC pretreated with SNP at 10-3 and 10-4 mol/L were 66.0%±2.6% and 52.9%±1.6% at 24 h, respectively. In the presence of ZnPP, however, they were 55.3%±1.2% and 43.3%±1.1%, respectively, which were lower than those in the absence of ZnPP (P<0.01).
Discussion
Proliferation and apoptosis of VSMC are 2 tightly linked pathways maintaining cellular homeostasis of vessel wall. The imbalance of VSMC proliferation and apoptosis is a cytological basis of vascular diseases under hypoxia. However, the regulatory mechanisms responsible for the disturbed proliferation and apoptosis of PASMC under hypoxia are still unclear.
Studies have demonstrated that eNOS-derived NO as well as other NOS isoforms derived NO can inhibit remodeling of the pulmonary vasculature induced by chronic hypoxia[4–7]. In our previous in vivo studies[19], we found that the plasma concentration of NO decreased and NO was involved in the remodeling of pulmonary arteries. In this study, we found that in PASMC exposed to hypoxia, the basal level of NO decreased markedly, and NO inhibited hypoxic PASMC proliferation and induced PASMC apoptosis, which attenuated pulmonary vascular remodeling, but the mechanisms by which NO regulated PASMC proliferation and apoptosis remains to be clarified.
It was reported that HO-1 was transcriptionally upregulated by hypoxia[20]. In the event that NO production was suppressed under hypoxic condition, gaseous CO, mainly produced by smooth muscle cells, became an important regulator of cell proliferation and apoptosis, evidenced by the significantly increased CO production and the increased expression of HO-1. Previous studies showed that hypoxia upregulates VSMC expression of the HO-1 gene, resulting in an increase in the production of CO, which in turn inhibits cellular proliferation[8–10]. In the present study, HO-1 mRNA levels showed a 2-fold increase at 12 h of hypoxia and started to decline by 24 h, with corresponding changes in CO production. Meanwhile, our study indicated that while PASMC proliferated in response to hypoxia, the reduction of CO production by treatment with ZnPP enhanced the proliferative response even further. Our findings are in agreement with those of Morita et al[8], who suggested that CO released by hypoxic PASMC was responsible for the inhibition of hypoxia-induced proliferation of PASMC, even though this response was transient. An interesting finding in this study was that endogenous CO induced PASMC apoptosis in a time-dependent manner, since the inhibition of CO production by ZnPP markedly decreased the number of apoptotic cells only when the cells were exposed to long-time hypoxia for 24 h. The underlying mechanism, however, was unknown. It was possible that cell-cell or cell-matrix interactions, which involved synthesis or release of growth factors, cytokines or biological messengers as NO and CO, maintained the vascular homeostasis.
Recent evidence suggested that NO was a potent inducer of the HO-1 gene expression in VSMC[11–16]. We demonstrated that HO-1 mRNA expression and CO production were shown to increase after administration of SNP, a donor of NO, in both normoxic and hypoxic cells. However, the mechanism by which NO induced HO-1 gene expression under both normoxic and hypoxic conditions is not entirely clear. Durante et al suggested that the cyclic guanosine monophosphate (cGMP) signaling pathway was not involved, since lipophilic analogues of cGMP failed to stimulate HO-1 expression, and the inhibitors of the cGMP breakdown or cGMP kinase did not affect NO-mediated HO-1 gene induction[12]. A previous report demonstrated that over-expression of HO-1 and the consequent elevation in heme oxygenase activity of endothelial cells exposed to hypoxia were associated with a transient formation of endogenous S-nitrosothiols due to an early induction of the induced nitric oxide synthase gene[21].
Thus, since both NO and CO have been shown to exert antiproliferative and pro-apoptotic effects on PASMC, it was reasonable to propose that the HO-1/CO pathway might play an important role in NO-regulated hypoxic pulmonary vascular structural remodeling. We hypothesized that NO and CO interact to regulate the proliferation and apoptosis of hypoxic PASMC. In this regard, we aimed to explore the role of CO in the regulation of hypoxic PASMC proliferation and apoptosis by NO. Our present study shows that in the presence of ZnPP, an inhibitor of HO, PCNA expressions in hypoxic PASMC pretreated with SNP were much higher than those in the absence of ZnPP at both 12 and 24 h (all P<0.01), suggesting that CO might be involved in a regulatory process where NO inhibits PASMC proliferation under hypoxic condition. In the presence of ZnPP, caspase-3 expressions of hypoxic PASMC pretreated with SNP were much lower than those in the absence of ZnPP at both 12 and 24 h (all P<0.01), indicating that CO might be involved in a regulatory process where NO promoted apoptosis under hypoxic condition. It is likely that the HO-1/CO pathway was responsible for modulating PASMC proliferation and apoptosis by NO. The induction of HO-1 activity and CO production by NO in PASMC may be of pathophysiological significance in regulating the proliferation and apoptosis of PASMC under both normoxic and hypoxic conditions.
In addition, our study found that caspase-3 might be involved in the signaling pathway of PASMC proliferation and apoptosis regulated by NO and CO, respectively. Previous studies have shown that NO induced vascular smooth muscle cell apoptosis via the activation of caspase-3[22,23]. Our findings of increased expression of caspase-3 in NO-induced apoptotic PASMC thus corroborated with those of other studies and provided a new insight of the mechanisms underlying hypoxic pulmonary vascular remodeling.
Our studies suggest that the production of NO was inhibited under hypoxic conditions, which again promoted the proliferation and suppressed the apoptosis of PASMC. However, there was a defensive pathway, HO-1/CO system, which was upregulated under hypoxia and might antagonize the effect of a decrease in NO. As NO and CO are both small molecules mainly produced by vascular endothelial cells and smooth muscle cells, respectively, when the production of NO was insufficient or impaired because of the injured endothelial cells under some stimulants, CO produced by smooth muscle cells might be increased to compensate.
In conclusion, CO was responsible for the regulation of VSMC growth similar to NO, and the HO-1/CO pathway is responsible for the suppression of smooth muscle cell proliferation and the promotion of apoptosis afforded by NO. Thus, HO-1 is an important cellular target of NO, and it was possible that CO compensated for the insufficiency or impairment of NO. It was possible that NO regulated the proliferation and apoptosis of VSMC by regulating the production of CO. Caspase-3 might be involved in the regulating pathway of PASMC proliferation and apoptosis. Whether there were other mechanisms involved in the regulation of PASMC proliferation and apoptosis by NO needs further study. Finally, our results might help to improve the understanding of the interactions of low molecular gas transmitters NO and CO as well as their pathophysiological signi-ficance.
References
- Kourembanas S, Morita T, Christou H, Liu YX. Hypoxic responses of vascular cells. Chest 1998;114:S25-8.
- Dzau VJ, Gibbons GH. Vascular remodeling: mechanisms and implications. J Cardiovasc Pharmacol 1993;21:S1-5.
- Gibbons GH, Dzau VJ. The emerging concept of vascular remodeling. N Engl J Med 1994;330:1431-8.
- Qi JG, Du JB, Jia JF. Endogenous nitric oxide regulates hypoxic pulmonary vascular remodeling in rats. Chin J Pediatr 1999;37:104-6.
- Mitani Y, Maruyama K, Sakurai M. Prolonged administration of L-Arginine ameliorates chronic pulmonary hypertension and pulmonary vascular remodeling in rats. Circulation 1997;96:689-97.
- Ozaki M, Kawashima S, Yamashita T, Ohashi Y. Reduced hypoxic pulmonary vascular remodeling by nitric oxide from the endothelium. Hypertension 2001;37:322-7.
- Smith JD, McLean SD, Nakayama DK. Nitric oxide causes apoptosis in pulmonary vascular smooth muscle cells. J Surg Res 1998;79:121-7.
- Morita T, Mitsialis SA, Koike H, Liu YX, Kourembanas S. Carbon monoxide controls the proliferation of hypoxic vascular smooth cells. J Biol Chem 1997; 272: 32 804–9.
- Shi Y, Du JB, Gong LM, Zeng CM, Tang XY, Tang CS. The regulating effect of heme oxygenase/carbon monoxide on hypoxic pulmonary vascular structural remodeling. Biochem Biophys Res Commun 2003;306:523-9.
- Zhen G, Xue Z, Zhang Z, Xu Y. Carbon monoxide inhibits proliferation of pulmonary smooth muscle cells under hypoxia. Chin Med J (Engl) 2003;116:1804-9.
- Hartsfield CL, Alam J, Cook JL, Choi AM. Regulation of heme oxygenase-1 gene expression in vascular smooth muscle cells by nitric oxide. Am J Physiol 1997;273:L980-8.
- Durante W, Kroll MH, Christodoulides N, Peyton KJ. Nitric oxide induces heme oxygenase-1 gene expression and carbon monoxide production in vascular smooth muscle cells. Circ Res 1997;80:557-64.
- Durante W, Christodoulides N, Cheng K, Peyton KJ. cAMP induces heme oxygenase-1 gene expression and carbon monoxide production in vascular smooth muscle. Am J Physiol 1997;273:H313-23.
- Liu Y, Christou H, Morita T, Laughner E. Carbon monoxide and nitric oxide suppress the hypoxic induction of vascular endothelial growth factor gene via the 5’enhancer. J Biol Chem 1998; 273: 15 257–62.
- Du JB, Yan H, Wei B, Li J, Qi JG, Tang CS. Effect of L-arginine on collagen of high flow-induced pulmonary arterial remodeling. Circ J 2005;69:603-8.
- Vanhoutte PM. Endothelial control of vasomotor function: from health to coronary disease. Circ J 2003;67:572-5.
- Shibahara S, Muller R, Taguchi H, Yoshida T. Cloning and expression of cDNA for rat heme oxygenase. Proc Natl Acad Sci 1985;82:7865-9.
- Chalmers AH. Simple sensitive measurement of carbon monoxide in plasma. Clin Chem 1991;37:1442-5.
- Qi J, Du J, Wang L, Zhao B, Tang C. Alleviation of hypoxic pulmonary vascular structural remodeling by L-arginine. Chin Med J (Engl) 2001;114:933-6.
- Lee PJ, Jiang BH, Chin BY, Iyer NV, Alam J. Hypoxia induced factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia. J Biol Chem 1997;272:5375-81.
- Motterlini R, Foresti R, Bassi R, Calabrese V, Clark JE, Green CJ. Endothelial heme oxygenase-1 induction by hypoxia. Modulation by inducible nitric-oxide synthase and S-nitrosothiols. J Biol Chem 2000; 275:13 613–20.
- Wang HE, Keiser JA. Molecular characterization of rabbit CPP32 and its function in vascular smooth muscle cells apoptosis. Am J Physiol 1998;43:H1132-40.
- Mallat Z, Ohan J, Leseche G, Tedgui A. Colocalization of CPP32 with apoptotic cells in human atherosclerotic plaques. Circulation 1997;96:424-8.