
Neuroprotection of selenite against ischemic brain injury through negatively regulating early activation of ASK1/JNK cascade via activation of PI3K/AKT pathway1
Introduction
Selenium, an essential biological trace element, has been shown to modulate functions of many regulatory proteins involved in signal transduction, and to affect a variety of cellular activities including cell growth, survival and death[1,2]. The relationship between selenium and signal molecules has not been well elucidated. There are a lot of suppositions in recent years. For example, it has been found that physio-logical concentrations of selenium (<3 µmol/L) reduces apo-ptosis signal regulating kinase 1 (ASK1) activity and induces the phosphatidylinositol 3-kinase (PI3K)/AKT pathway in HT1080 cells[3–6]. It has been shown that selenium compounds such as selenite (commercially available as sodium selenite) has greater than 80% bioavailability. Selenite increases cell proliferation through the upregulation of Bcl-2 expression, mitochondrial membrane potential, ATP generation, and glucose uptake mediated by PI3K pathway. Selenite, as an antioxidant, can also block apoptosis induced by H2O2, which is related to blocking ASK1 activity and further stimulating the activation of the PI3K/AKT pathway[6,7].
The cellular decision to undergo apoptosis is determined by the integration of multiple survival and death signals[8]. PI3K/AKT is a major cell survival pathway that has been extensively studied recently[9]. AKT [also known as protein kinase-B(PKB)] represents a subfamily of serine/threonine kinase. Three members of this family including AKT1, AKT2 and AKT3, have been identified so far. AKT is activated in a PI3K-dependented manner by a variety of stimuli, including growth factors, protein phosphatase inhibitors, and stress[10–12]. Phosphorylation of residues Thr-308 and Ser-473 is required for AKT activity. Several targets of AKT that have been identified have roles in the regulation of apoptosis, such as the pro-apoptotic proteins Bcl-2/Bcl-xL-associated death protein (BAD) and caspase-9 and forkhead transcription factor like 1(FKHRL1). Activation of AKT blocks BAD binding to Bcl-x, inhibits caspase-9 protease activity, and blocks FKHRL1 function, reducing Fas ligand transcription, and thereby inducing anti-apoptotic effects[13].
ASK1 is a member of the mitogen-activated protein kinase (MAPK) family that activates both the mitogen-activated protein kinase kinase(MKK)4/MKK7-c-jun N-terminal kinase (JNK) and MKK3/MKK6-p38 signaling cascades[14,15]. ASK1 is a general mediator of cell death in response to a variety of stimuli, including oxidative stress and chemotherapeutic drugs such as cisplatin and paclitaxel[16,17]. Oxidative stress induces phosphorylation of Thr-845 in the activation loop of ASK1, which is correlated with ASK1 activity and ASK1-dependent apoptosis[3,5]. On the other hand, AKT1 and ASK1 are physically associated; the phosphorylation of ASK1 at Ser-83 is a consensus AKT1 phosphorylation site. Phosphorylation at Ser-83 by AKT1 is related to a decrease in stimulated ASK1 kinase activity and promotes cell survival[13].
JNK, a member of MAPK, is important in inducing neuronal death. The JNK are encoded by 3 genes: JNK1, JNK2 and JNK3. Recent studies show that JNK is involved in apoptosis. For instance, overexpression of a constitutively activated JNK kinase potentiates apoptosis of PC12 cell in response to nerve growth factor deprivation[18]. Activated JNK, in turn, phosphorylates a number of transcription factors, especially the c-Jun of component of AP-1, and cellular proteins, particularly those associated with apoptosis (for example, Bcl-2, P53)[19].
In the present study, we aimed to investigate whether selenite, a known antioxidant, could decrease the activation of the ASK1/JNK signaling cascade in cerebral ischemia/reperfusion by activating the PI3K/AKT pathway in rat hippocampi, and the neuroprotective effect of selenite against ischemic injury.
Materials and methods
Animal surgical procedures We selected adult male Sprague-Dawley (SD) rats weighing 250–300 g (Shanghai Experimental Animal Center, Chinese Academy of Science, Shanghai, China). All rats were divided into the following groups: sham group, ischemia/reperfusion group, saline-treated group, saline+DMSO-treated group, selenite-treated group and selenite+LY294002-treated groups. Every group was derived from 6 independent animals. The experimental procedures were approved by the local legislation for ethics of experiments on animals. Cerebral ischemia was induced by 4-vessel occlusion as described before[20]. Briefly, the rats were anesthetized with chloral hydrate (350 mg/kg, ip). The bilateral vertebral arteries were occluded by electro-cautery. On the following day, the bilateral carotid arteries were occluded for specified time using aneurysm clips. During ischemia, the animals were required to match the following criteria: completely flat electroencephalo-graphs, maintenance of dilated pupils, absence of a cornea reflex when exposed to strong light stimulation, and maintenance of rectal temperature at about 37 ºC. Those not matching these criteria or with seizures were all excluded. The sham operation was performed using the same surgical exposure procedures except for the occlusion of the carotid artery.
Brain tissues and drug treatment To test the time courses of the expression and phosphorylation of AKT1 and ASK1 after I/R, the rats were decapitated at 0, 30 min, 3, 6, 12 h, 1 and 3 d after 15 min of ischemia. Next, to evaluate the specific mechanism of the relationship between the PI3K/AKT and ASK1/JNK pathways, an antioxidant selenite, and LY294002, a specific inhibitor of the PI3K were used. The rats were pretreated only with selenite (0.3 mg/kg dissolved in 0.9% saline; Sigma, St Louis, MO, USA) every 24 h for 7 d by means of intravenous injection of the tail or combined with LY294002 from d 5 by left cerebral ventricle injection before surgery. LY294002 was dissolved in DMSO and was used by left cerebral ventricle injection (25 mmol/L, 5 µL, from the bregma: anteroposterior, -0.8 mm; lateral, 1.5 mm; depth, 3.5 mm). The control rats received equal volumes of 0.9% saline or DMSO. When the rat brains were removed by dissection, the hippocampi were immediately frozen in liquid nitrogen. All samples were stored at -80 °C until use.
Tissue preparation The hippocampi tissues were homogenized in ice-cold homogenization buffer (50 mmol/L Mops pH 7.4, 100 mmol/L KCl, 50 mmol/L NaF, 20 mmol/L sodium pyrophosphate, 20 mmol/L β-phosphogrycerol, 320 mmol/L sucrose, 0.2 mmol/L DTT, 1 mmol/L edetic acid, 1 mmol/L egtazic acid, 0.5 mmol/L MgCl2, 1 mmol/L Na3VO4, 1 mmol/L p-nitrophenyl phosphate and protease inhibitors: 5 µg/mL aprotinin, 5 µg/mL leupeptin, 5 µg/mL pepstatin A, 1 mmol/L phenyl-methylsulfonyl fluoride, and 1 mmol/L benzamidine). Then the homogenates were centrifuged at 800×g for 15 min at 4 °C. The supernatants were collected and protein concentration was determined by the method of Lowry. Samples were stored at -80 °C until use.
Western blotting analysis The samples were separated by 10% or 7.5% SDS-PAGE and electrotransferred onto nitrocellulose membranes by the method described before[20]. After blocking with 3% bovine serum albumin (BSA) for 3 h, the membrane was probed with proper primary antibodies at 4 °C overnight. Bound antibodies were detected by using alkaline phosphatase conjugated goat anti-mouse IgG or goat anti-rabbit IgG (Sigma, USA) for 2 h. Immunoreactivity was detected by a NBT/BCIP assay kit (Promega, Madison, WI, USA) according to the manufacturer’s instructions. The bands on the membranes were scanned and analyzed with an image analyzer (LabWorks Software; UVP Inc, Upland, CA, USA).
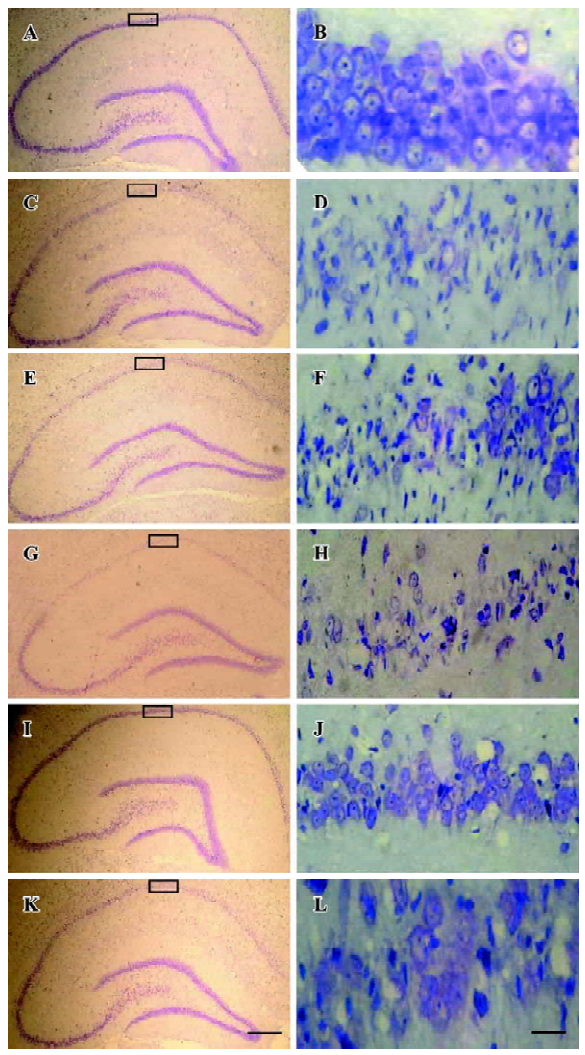
Histology The rats were perfusion-fixed with 4% paraformaldehyde in 0.1 mol/L sodium phosphate buffer (pH 7.4) under anesthesia after 3 d of I/R. The brains were removed quickly and further fixed with the same fixation solution at 4 °C overnight. The post-fixed brains were embedded by paraffin, followed by preparation of coronal sections 5 μm thick using a microtome. The paraffin-embedded brain sections were deparaffinized with xylene and rehydrated by ethanol at graded concentrations of 100%–70% (v/v), followed by washing with water. The sections were stained with 0.1% (w/v) cresyl violet, and were examined with light microscopy. The number of surviving hippocampal CA1 pyramidal cells per 1 mm length was counted as the neuronal density[21].
Antibodies Rabbit polyclonal anti-p-AKT1 (#9271) was obtained from Upstate Biotechnology (Lake Placid, NY, USA). Rabbit polyclonal anti-p-ASK1-Ser83 (#3761), rabbit polyclonal anti-p-ASK1-Thr845 (#3765), rabbit polyclonal anti-ASK1 (#3762), and rabbit polyclonal anti-p-MKK4 (#9151) were purchased from Cell Signaling Technology (Danvers, MA, USA). Rabbit polyclonal anti-MKK4 (sc964) was obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Rabbit polyclonal anti-p-JNK1/2 was obtained from Promega Corporation (USA). Rabbit polyclonal anti-JNK1/2 was obtained from Sigma Biotechnology (USA). Mouse polyclonal anti-AKT1 (sc1618) was obtained from Santa Cruz Biotechnology (USA). The secondary antibodies used in our experiment were goat anti-mouse IgG, goat anti-rabbit IgG, and were from Sigma (USA).
Statistical analysis Values were expressed as mean±SD from 6 independent rats. Statistical analysis of the results was performed by one-way analysis of variance followed by Duncan’s new multiple range method. P-values<0.05 were considered significant.
Results
Effects of selenite and LY294002 on AKT1 activation after I/R To explore the effects of selenite and LY294002 on the activation of the PI3K/AKT pathway, we initially examined the activation and expression of AKT1 at 0, 30 min, 3, 6, 12 h, 1 and 3 d of reperfusion following 15 min of ischemia. AKT1 activation first decreased at 0 min of reperfusion, then increased rapidly compared with the control from 30 min of reperfusion, and decreased generally from 6 h before returning to the control level at 1 d. Total AKT1 protein levels were unchanged (Figure 1A, 1B). Selenite strengthened AKT1 activation at 30 min of reperfusion compared with the vehicle control, whereas the combined use of LY294002 blocked the increase of AKT1 activation induced by pretreatment only with selenite. The protein levels of AKT1 were unaffected (Figure 1C, 1D).
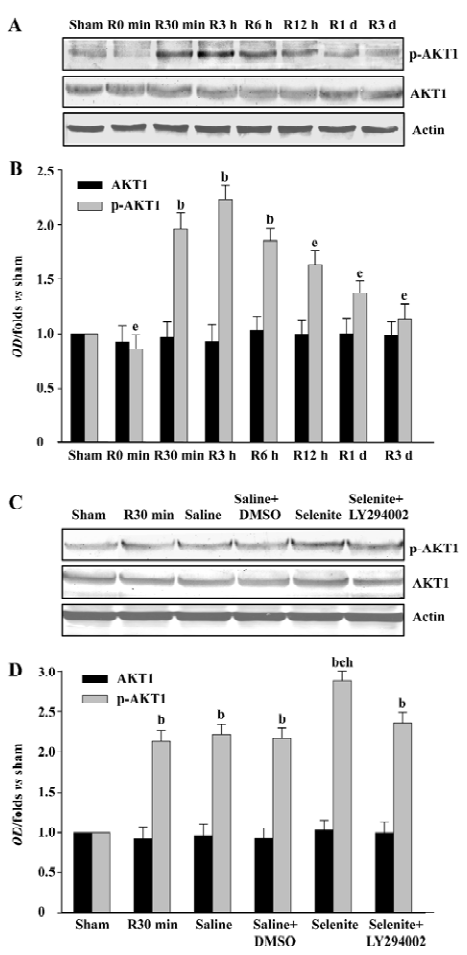
Effects of selenite and LY294002 on ASK1-Ser-83 phosphorylation after I/R The phosphorylation of ASK1 at Ser-83 is a consensus AKT1 phosphorylation site. To examine whether selenite and LY294002 had the same effects on the Ser-83 phosphorylation of ASK1 as that of AKT1, samples from rat hippocampi at various time points of reperfusion following 15 min of ischemia were also used. The Ser-83 phosphorylation of ASK1 had a similar tendency as that of AKT1, except that the phosphorylation of AKT1 and ASK1 peaked at 3 h and 6 h of reperfusion, respectively. The expression of ASK1 was not affected (Figure 2A, 2B). Pretreatment with selenite significantly increased the Ser-83 phosphorylation of ASK1 at 30 min of reperfusion compared with the vehicle control, whereas the combined use of both selenite and LY294002 blocked the increase of the Ser-83 phosphorylation of ASK1 induced by pretreatment only with selenite. Meanwhile, selenite or LY294002 had no effect on the expression of ASK1 (Figure 2C, 2D).
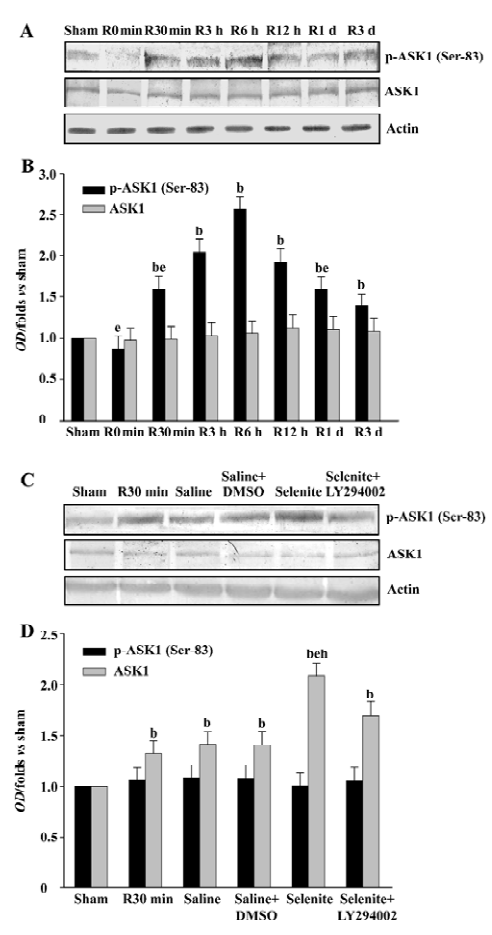
Effects of selenite and LY294002 on ASK1-Thr-845 phosphorylation after I/R The increased Ser-83 phosphorylation induced by selenite might represent the decreased ASK1 activation. Thus, we next examined the possible effect of selenite and LY294002 on Thr-845 phosphorylation, which is correlated with ASK1 activity. ASK1 was phosphorylated at Thr-845 with 2 peaks which occurred at 3 h and 3 d of reperfusion, respectively, whereas total protein levels of ASK1 were unaffected (Figure 3A, 3B). At the 30 min time point, selenite obviously decreased the Thr-845 phosphorylation of ASK1 compared with the vehicle control. However, pretreatment with both selenite and LY294002 restored the phenomena induced by pretreatment only with selenite. The protein levels of ASK1 were not affected (Figure 3C, 3D).

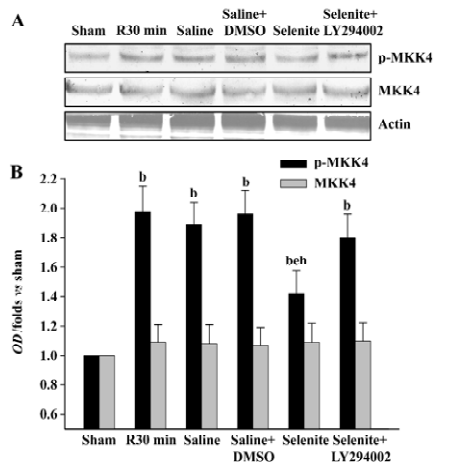
Effects of selenite and LY294002 on MKK4 activation after I/R We also examined the effects of selenite and LY294002 on the activation of MKK4, a downstream substrate of ASK1 at the 30 min time point of reperfusion. Selenite significantly inhibited MKK4 activation at 30 min. On the contrary, the combined use of LY294002 reversed the decrease of MKK4 activation induced by pretreatment only with selenite. The expression of MKK4 was unchanged (Figure 4).
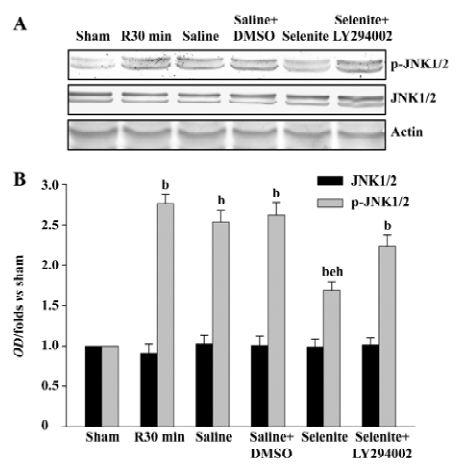
Effects of selenite and LY294002 on JNK1/2 activation after I/R Finally, we examined whether selenite could inhibit the activation of JNK1/2, a downstream substrate of MKK4, and whether LY294002 could reverse the effect of selenite. The earlier active peak of JNK1/2 at 30 min of reperfusion was selected. Selenite obviously inhibited JNK1/2 activation at 30 min of reperfusion. On the contrary, LY294002 blocked the decreased JNK1/2 activation at 30 min of reperfusion induced by pretreatment only with selenite. The protein levels of JNK1/2 were unaffected (Figure 5).
Neuroprotective effects of selenite against I/R-induced neuronal loss in hippocampal CA1 regions To explore the neuroprotective effects of selenite against I/R-induced neuronal loss, cresyl violet staining was used to examine the survival of CA1 pyramidal neurons. Normal cells showed round and pale-stained nuclei. The shrunken cells with pyknotic nuclei after ischemia were counted as dead cells. Transient brain ischemia, followed by 3 d of reperfusion, induced severe cell death. Pretreatment with selenite (0.3 mg/kg) for 7 d before ischemia significantly decreased neuronal degeneration, while pretreatment with LY294002 and selenite aggravated cell death compared with only selenite pretreatment. However, the saline and DMSO-treated groups had no obvious neuroprotective effects against I/R-induced neuronal loss in hippocampal CA1 regions (Figure 6, Table 1).
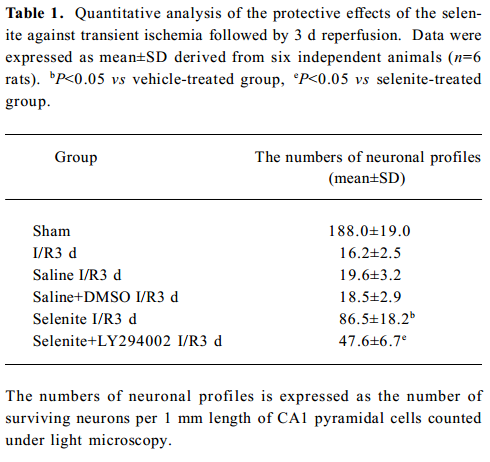
Full table
Discussion
Recent studies have illustrated that the ischemia insult can induce the generation of free radicals, and the increase in free radicals becomes larger and unambiguous especially during early reperfusion[22]. Moreover, free radicals such as H2O2 which are formed in association with a variety of oxidative stress-induced disorders may be related to cell death and hence play an important role in apoptosis. The brain is particularly susceptible to damage due to oxidative stress because neurons are rich in polyunsaturated fatty acids, and levels of endogenous antioxidant enzymes in neuronal tissue are low[23,24]. Therefore, oxidative stress may contribute to neuronal cell death due to ischemia and reperfusion. Several synthetic free radical scavengers have been evaluated in animal models of cerebral ischemia and reperfusion and have been shown to be protective. For example, antioxidants, N-acetylcysteine (NAC) and pyrrolidine dithiocarbamate have important neuroprotective effects against I/R-induced injury through downregulating the activation of NF-κB[25,26,27]. Therefore, our present study is designed to further investigate the protective effects of selenite, an antioxidant, during reperfusion after 15 min of global brain ischemia in rat hippocampi. Our results suggest that the pretreatment with selenite (0.3 mg/kg) for 7 d before ischemia can strongly decrease neuronal degeneration compared with the control rats.
Previous papers have shown that reactive oxidative species (ROS) such as H2O2 can induce the activation of the ASK1/JNK signaling cascade[6]. As a known oxidative stress-responsive SEK1 kinase, ASK1-mediated apoptosis is associated with ROS[20]. Moreover, we have found that the early JNK activation was also associated with ROS following ischemia, and the activation of ASK1 and JNK can be obviously inhibited by antioxidants, such as NAC[20,28]. Further-more, it has been reported that, as an anti-apoptotic signal molecule, AKT is activated by H2O2 which is associated with a much shorter duration. Recent studies have demonstrated that selenite blocks oxidative stress-induced apoptosis through the activation of the PI3K/AKT pathway and the inhibition of the ASK1/JNK pathway in HT1080 cells[6]. Thus, in our present study, an antioxidant selenite was used to test the relationship between the signal molecules and oxidative stress. As we expected, selenite strongly inhibited the activation of ASK1/JNK cascade and simultaneously increased the AKT1 activation. Together with previous studies, our results show that antioxidants can block the activation of the ASK1/JNK cascade and further stimulate PI3K/AKT activities during early reperfusion after cerebral ischemia, which also implies that there might be some delicate balance between the pro-apoptotic ASK1/JNK cascade and the anti-apoptotic PI3K/AKT pathway.
It is well known that AKT is a critical mediator of cell survival, and the prolonged activation of AKT is necessary for the prevention of apoptosis. Compared with AKT, ASK1 plays a central role in the mechanisms of stress- and cytokine-induced apoptosis. ASK1 is composed of an inhibitory N-terminal domain, an internal kinase domain and a C-terminal regulatory domain, which is a central target of many cellular survival factors that bind to its different domains to keep it in an inactive state. It has been reported that AKT and ASK1 are physically associated. ASK1 can be phosphorylated at several sites, and these phosphorylation sites regulate ASK1 activity in both positive and negative manners. For example, phosphorylation at Ser-967 is essential for ASK1 associated with the 14-3-3 protein and suppression of cell death[29]. Oxidative stress induces dephosphorylation of Ser-967 and phosphorylation of Thr-845 in the activation loop of ASK1, and both are correlated with ASK1 activation and ASK1-dependent apoptosis[30,31]. Conversely, several serine/threonine protein kinases such as AKT1 directly phosphorylates ASK1 at Ser-83, a consensus AKT1 phosphorylation site, which attenuates ASK1 activity and promotes cell survival[32]. That is to say, the activation of the PI3K/AKT pathway can reduce the activation of the ASK1/JNK signaling cascade in a manner dependent on phosphorylation of Ser-83 of ASK1. Thus, in our present study, LY294002, a specific PI3K inhibitor was used to confirm the inhibitory mechanism of the PI3K/AKT pathway on the ASK1/JNK cascade during early reperfusion after cerebral ischemia. Fantastically, the inhibition of AKT1 by the PI3K inhibitor significantly reversed the activation of the ASK1/JNK cascade as our data shown. Although the exact mechanism by which AKT1 inhibits ASK1 was not clear, it provided evidence to support the fact that AKT1 negatively regulated ASK1 activity by phosphorylating ASK1 at Ser-83 in response to ischemia stress.
In conclusion, our results clearly demonstrated that antioxidant selenite could increase the activation of the anti-apoptotic PI3K/AKT pathway and decrease the activation of the pro-apoptotic ASK1/JNK cascade via phosphorylating ASK1 at Ser-83 residue by AKT1 during early reperfusion after cerebral ischemia in rat hippocampi. It gave us a novel clue that antioxidants might suppress oxidative stress-induced ischemic brain insult through the activation of the PI3K/AKT pathway and the inhibition of the ASK1/JNK cascade.
References
- Park HS, Park E, Kim MS, Ahn K, Kim IY, Choi EJ. Selenite inhibits the c-Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK) through a thiol redox mechanism. J Biol Chem 2000;275:2527-31.
- Lee YC, Tang YC, Chen YH, Wong CM, Tsou AP. Selenite-induced survival of HuH7 hepatoma cells involves activation of focal adhesion kinase-phosphatidylinositol 3-kinase-Akt pathway and Rac1. J Biol Chem 2003;278:39615-24.
- Baum MK, Shor-Posner G, Lai S, Zhang G, Lai H, Fletcher MA, et al. High risk of HIV-related mortality is associated with selenium deficiency. J Acquir Immune Defic Syndr Hum Retrovirol 1997;15:370-4.
- Ganther HE. Selenium metabolism, selenoproteins and mechanisms of cancer prevention: complexities with thioredoxin reductase. Carcinogenesis 1999;20:1657-66.
- Rayman MP. The importance of selenium to human health. Lancet 2000;356:233-41.
- Yoon SO, Kim MM, Park SJ, Kim D, Chung J, Chung AS. Selenite suppresses hydrogen peroxide-induced cell apoptosis through inhibition of ASK1/JNK and activation of PI3-K/Akt pathways. Faseb J 2002;16:111-3.
- Gupta R, Singh M, Sharma A. Neuroprotective effect of antioxidants on ischaemia and reperfusion-induced cerebral injury. Pharmacol Res 2003;48:209-15.
- Kim AH, Khursigara G, Sun X, Franke TF, Chao MV. Akt phosphorylates and negatively regulates apoptosis signal-regulating kinase 1. Mol Cell Biol 2001;21:893-901.
- Cantley LC. The phosphoinositide 3-kinase pathway. Science 2002;296:1655-7.
- Franke TF, Yang SI, Chan TO, Datta K, Kazlauskas A, Morrison DK, et al. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell 1995;81:727-36.
- Liu AX, Testa JR, Hamilton TC, Jove R, Nicosia SV, Cheng JQ. AKT2, a member of the protein kinase B family, is activated by growth factors, v-Ha-ras, and v-src through phosphatidylinositol 3-kinase in human ovarian epithelial cancer cells. Cancer Res 1998;58:2973-7.
- Shaw M, Cohen P, Alessi DR. The activation of protein kinase B by H2O2 or heat shock is mediated by phosphoinositide 3-kinase and not by mitogen-activated protein kinase-activated protein kinase-2. Biochem J 1998;336:241-6.
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999;96:857-68.
- Nishitoh H, Saitoh M, Mochida Y, Takeda K, Nakano H, Rothe M, et al. ASK1 is essential for JNK/SAPK activation by TRAF2. Mol Cell 1998;2:389-95.
- Yuan ZQ, Feldman RI, Sussman GE, Coppola D, Nicosia SV, Cheng JQ. AKT2 inhibition of cisplatin-induced JNK/p38 and Bax activation by phosphorylation of ASK1: implication of AKT2 in chemoresistance. J Biol Chem 2003; 278: 23 432–40.
- Gotoh Y, Cooper JA. Reactive oxygen species- and dimerization-induced activation of apoptosis signal-regulating kinase 1 in tumor necrosis factor-alpha signal transduction. J Biol Chem 1998;273:17477-82.
- Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, et al. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. Embo J 1998;17:2596-606.
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 1995;270:1326-31.
- Manning AM, Davis RJ. Targeting JNK for therapeutic benefit: from junk to gold? Nat Rev Drug Discov 2003;2:554-65.
- Zhang Q, Zhang G, Meng F, Tian H. Biphasic activation of apoptosis signal-regulating kinase 1-stress-activated protein kinase 1-c-Jun N-terminal protein kinase pathway is selectively mediated by Ca2+-permeable alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptors involving oxidative stress following brain ischemia in rat hippocampus. Neurosci Lett 2003;337:51-5.
- Guan QH, Pei DS, Zhang QG, Hao ZB, Xu TL, Zhang GY. The neuroprotective action of SP600125, a new inhibitor of JNK, on transient brain ischemia/reperfusion-induced neuronal death in rat hippocampal CA1 via nuclear and non-nuclear pathways. Brain Res 2005;1035:51-9.
- Piantadosi CA, Zhang J. Mitochondrial generation of reactive oxygen species after brain ischemia in the rat. Stroke 1996;27:327-32.
- Juurlink BH, Sweeney MI. Mechanisms that result in damage during and following cerebral ischemia. Neurosci Biobehav Rev 1997;21:121-8.
- Sugawara T, Chan PH. Reactive oxygen radicals and pathogenesis of neuronal death after cerebral ischemia. Antioxid Redox Signal 2003;5:597-607.
- Carroll JE, Howard EF, Hess DC, Wakade CG, Chen Q, Cheng C. Nuclear factor-kappa B activation during cerebral reperfusion: effect of attenuation with N-acetylcysteine treatment. Brain Res Mol Brain Res 1998;56:186-91.
- Sekhon B, Sekhon C, Khan M, Patel SJ, Singh I, Singh AK. N-Acetyl cysteine protects against injury in a rat model of focal cerebral ischemia. Brain Res 2003;971:1-8.
- Shen WH, Zhang CY, Zhang GY. Antioxidants attenuate reperfusion injury after global brain ischemia through inhibiting nuclear factor-kappa B activity in rats. Acta Pharmacol Sin 2003;24:1125-30.
- Tian H, Zhang G, Li H, Zhang Q. Antioxidant NAC and AMPA/KA receptor antagonist DNQX inhibited JNK3 activation following global ischemia in rat hippocampus. Neurosci Res 2003;46:191-7.
- Zhang R, Luo D, Miao R, Bai L, Ge Q, Sessa WC, et al. Hsp90-Akt phosphorylates ASK1 and inhibits ASK1-mediated apoptosis. Oncogene 2005;24:3954-63.
- Tobiume K, Saitoh M, Ichijo H. Activation of apoptosis signal-regulating kinase 1 by the stress-induced activating phosphorylation of pre-formed oligomer. J Cell Physiol 2002;191:95-104.
- Goldman EH, Chen L, Fu H. Activation of apoptosis signal-regulating kinase 1 by reactive oxygen species through dephosphorylation at serine 967 and 14-3-3 dissociation. J Biol Chem 2004;279:10442-9.
- Milanesio M, Artioli G, Gualtieri AF, Palin L, Lamberti C. Template burning inside TS-1 and Fe-MFI molecular sieves: an in situ XRPD study. J Am Chem Soc 2003;125:14549-58.