
Cytogenetic and molecular genetic alterations in hepatocellular carcinoma
Introduction
Hepatocellular carcinoma (HCC) is one of the most common human malignant neoplasms, with a particularly high incidence in Chinese and African populations[1]. In 2000, HCC ranked as the fifth most frequent cancer, but the third leading cause of cancer death worldwide[2]. Unlike other cancers, the main causative agents for HCC, hepatitis B virus (HBV), hepatitis C virus (HCV) and aflatoxin (AFB), are well studied. Yet little is known about the molecular pathogenesis of HCC[3]. In fact, the majority of HCC are associated with a background of chronic liver disease. Therefore, hepatocarcinogenesis is believed to be a long-term process that involves multiple genetic alterations.
Recurrent chromosome alterations
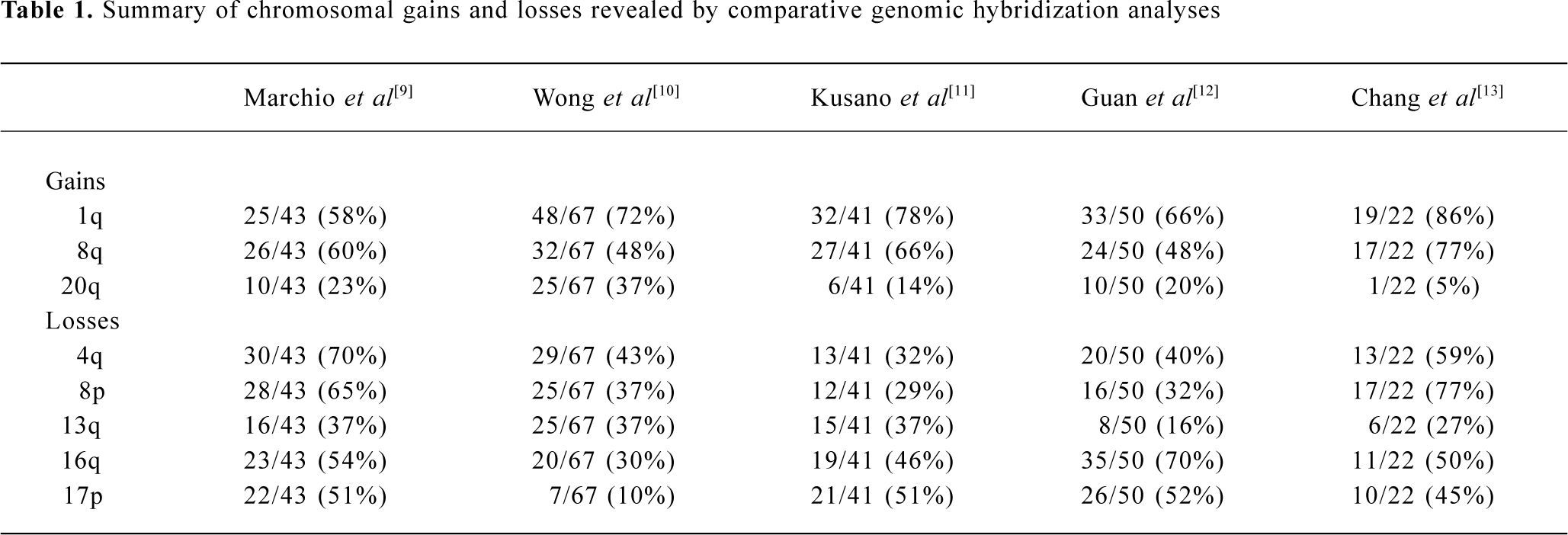
Chromosome aberrations are a hallmark of solid tumors and it has been known for decades that chromosome rearrangements exist in most if not all human tumors[4]. Addi-tionally, cytogenetic study followed by molecular analysis of recurring chromosome changes has greatly facilitated the identification of crucial oncogenes and tumor suppressors[5]. For example, the tumor suppresor gene Rb was identified based on the observation of chromosome deletion del(13)(q14) in retinoblastoma[6] and the proto-oncogene myc was shown to be involved in the chromosome translocation t(8;14) in human Burkett’s lymphoma[7]. The conventional comparative genomic hybridization (CGH) method provides a powerful means of global analysis of segmental chromosome gains or losses[8]. Since it was developed, CGH has been widely applied to the detection of recurrent chromosomal alterations in various cancers, including HCC[9–13]. Frequent non-random chromosomal gains and losses detected by CGH are summarized in Table 1, including gains of 1q, 6p, 8q, 17q, and 20q, and losses of 1p, 4q, 5q, 6q, 8p, 9p, 10q, 13q, 16q, 17p, 19p, and 22q[9–13]. In addition, the loss of heterozygosity (LOH) assay is used to define chromosomal regions with allelic deletions, and results revealed that LOH was frequently detected in 1p, 4q, 6q, 8p, 13q, 16q, and 17p[14–16]. These studies suggest the presence of multiple oncogenes or tumor suppressor genes in regions of recurrent gain or loss, respectively.
Full table
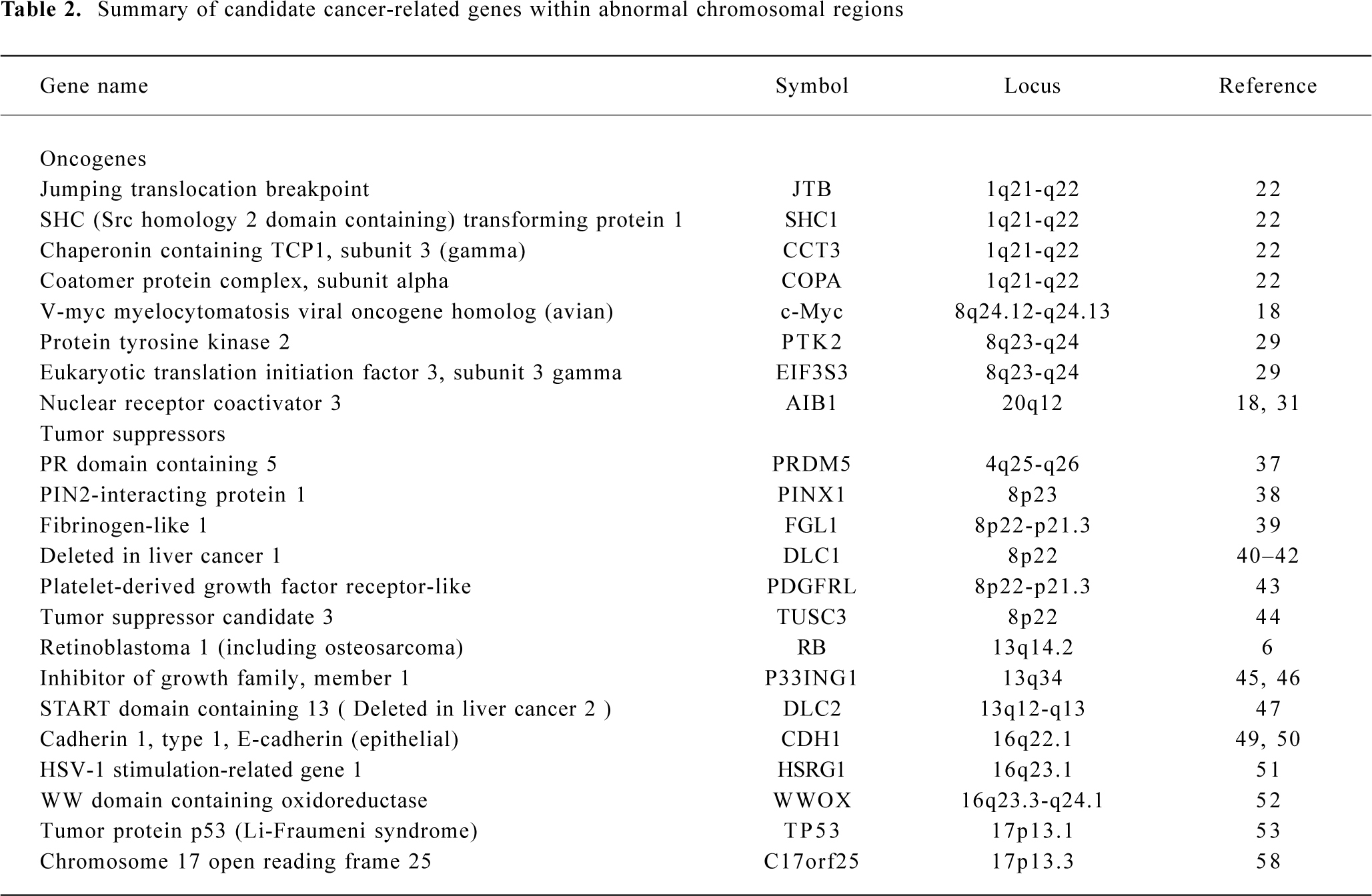
To avoid overlap with comprehensive reviews that have been published recently[3,17], this review will mainly focus on chromosomal gains on 1q, 8q, and 20q, and losses on 4q, 8p, 13q, 16q, and 17p in HCC. For simplicity, gene symbols will be used for all genes described throughout the review (details for all genes are summarized in Table 2).
Full table
Chromosome gains
Gain of 1q Gain of 1q is one of the most frequently detected alterations in HCC and has been suggested as an early genomic lesion in the process of HCC development[18], although the target oncogene within the amplified region has not been identified. CGH studies showed that the gain of 1q was detected in 58%–86% of HCC cases[9–13], with about 10% high-copy number amplification[12]. Several minimal amplifying regions (MAR) were mapped including 1q12-q22[12,19], 1q23.3-q25.3[20] and 1q24.2-1q43[21]. In an attempt to identify candidate oncogene(s) at 1q12-q22, Wong et al examined the expression level of several candidate onco-genes in this region using reverse transcription-polymerase chain reaction (RT-PCR). Significant overexpression of 4 genes known as JTB, SHC1, CCT3, and COPA were observed in tumors as compared with paired adjacent liver tissues[22].
Gain of 8q Gain of 8q is frequently observed in HCC (48%–77%)[9–13]. The gain of 8q is also frequently detected in many other solid tumors including those of the prostate[23], lung[24], esophagus[25], nasopharynx[26], ovary[27] and breast[28]. Amplification and overexpression of the c-myc oncogene has been reported in various solid tumors, including HCC. In our recent study, amplification of c-myc was correlated with HCC tumor size, tumor metastasis and recurrence. Amplification of c-myc was found in 11/13 (85%), 9/25 (36%) and 4/12 (33%) cases of large HCC (>9 cm), medium HCC (4–9 cm) and small HCC (<4 cm), respectively. In addition, a higher frequency of c-myc amplification was detected in metastatic tumors (45%) than in their primary HCC (29%), and in recurrent tumors (60%) than in their primary HCC (38%)[18]. Because the gain of 8q involved the entire long arm in this study, it is highly likely that one or more novel oncogenes are involved in HCC development apart from c-myc. In a recent report, Okamoto et al suggested 2 candidate oncogenes as amplification targets at 8q23-q24 known as PTK2 and EIF3S3. PTK2 encodes focal adhesion kinase, while EIF3S3 encodes the p40 subunit of eukaryotic translation initiation factor 3. Overexpression of these genes may be involved in HCC progression[29].
Gain of 20q Gain of 20q was detected in 3/5 CGH studies with a frequency of 20%–37% [9,10,12]. Gain of 20q was also frequently detected in breast cancer, and a steroid receptor coactivator, AIB1, has been identified within the commonly amplified region at 20q12[30,31]. Interestingly, gain of 20q is significantly associated with clinical stages and tumor size of HCC. Guan et al reported that gain of 20q occurred in 10/40 (25%) of stage II and III HCC but in 0/10 stage I HCC[12]. In addition, Wong et al reported that significantly more 20q gain was found in non-cirrhotic HCC. Strikingly, gain of 20q was observed in 9/12 (75%) non-cirrhotic, while only in 16/55 (29%) cirrhotic HCC (P=0.003)[10]. In our recent study, amplification of the AIB1 gene was correlated with HCC metastasis and recurrence. The amplification frequency of AIB1 was significantly higher in metastatic HCC (41%) than in matched primary HCC (23%), as well as being higher in recurrent HCC (60%) than in matched primary HCC (29%) (P < 0.05)[18].
Chromosome losses
Loss of 4q Loss of chromosome 4q was detected by CGH in 32%–70% of HCC cases[9–13]. Deletion of 4q has been well described because of its specificity for HCC. Several minimal deletion regions (MDR) on 4q have been detected by LOH including 4q12-q23, 4q13.1-q21.23, 4q13-q34, 4q21-q25, 4q24-q28, 4q28.2-q34.3, and 4q35[32–34]. In addition, Yeh et al reported recently the mapping of a 17.5 cM MDR between D4S1534 and D5S1572 by screening 149 HCC: a few candidate genes were found to be downregulated[35]. Biological evidence for tumor suppressor genes on 4q comes from a study that reversed the tumorigenic phenotype of a teratoma cell line by introducing a normal chromosome 4 into malignant teratoma cells using microcell-mediated chromosome transfer[36]. To date, candidate tumor suppressor genes correlated with 4q loss have remained unidentified. A recent report by Deng et al described the role of PRDM5 at 4q26 as a suppressor in various tumors including HCC. PRDM5 encodes a protein belonging to the PR-domain protein family. Epigenetic silencing was evident for this gene and its overexpression resulted in proapoptotic and growth suppressive functions[37].
Loss of 8p Loss of 8p is well documented in HCC with a frequency of 29%–77%[9–13]. By analyzing 50 HCC cases, we observed a correlation of 8p loss with advanced stage of disease and tumor size; most strikingly, 8p loss was present in 8/13 (62%) of large, 7/25 (28%) of medium, but only 1/12 (8%) of small HCC cases (P=0.014)[12]. Furthermore, 8p deletion was associated with HCC metastasis, suggesting the presence of one or more candidate gene(s) are involved in HCC progression[19]. Using the high-resolution LOH strategy, Jou et al reported that 50% of HCC showed LOH in chromosome 8p and the percentages of 3 MDR at 8p23.3-p23.1 (D8S504–D8S277), 8p22-p21.3 (D8S1106–D8S258) and 8p21.3-p12 (D8S258–D8S283) were 71%, 69%, and 63%, respectively[34]. Several candidate tumor suppressors have been isolated within these regions, including PINX1 at 8p23[38], FGL1 at 8p22[39] and DLC1[40] at 8p22-p21.3 regions. In the 8p23 region, Liao et al observed the under-expression of PINX1 (LPTS) in HCC cells and tissues, while ectopic expression of this gene in liver cancer cells SMMC-7721 resulted in growth suppression[38]. At 8p22, Yan et al reported reduced or undetectable expression of FGL1 in HCC speci-mens, and found a significant correlation with tumor differentiation[39]. Interestingly, Wong et al reported the significant under-expression of DLC1 mRNA and promoter hyper-methylation was found in 24% of primary HCC cases, suggesting a role of epigenetic silencing in DLC1 downregula-tion[41]. DLC-1 belongs to the rho family GTPase-activating proteins (RhoGAP) specific to RhoA and cdc42, which are implicated in cell migration control. Re-expression of DLC1 in HCC cells resulted in caspase-3-dependent apoptosis, inhibition of cell growth and invasiveness in vitro, and reduction of tumor formation in nude mice[42]. Furthermore, candidate tumor suppressors known as PDGFRL[43] and TUSC3[44] were identified in the 8p22-p21.3 region.
Loss of 13q Loss of 13q was detected in 16%–37% of HCC cases[9–13]. LOH analyses also showed a high percentage of allelic deletion on 13q (40%–55%)[14–16]. Jou et al identified 5 MDR with high frequencies of LOH including 13q12.11-q12.3 (71%), 13q13.1-q22.1 (79%), 13q31.3-q32.2 (67%), 13q32.2-q33.3 (73%) and 13q33.3-q34 (75%)[34]. MDR in 13q13.1-q22.1 (D13S171–D13S156) showed the highest frequency of LOH, and well-documented tumor suppressors including BRCA2 and RB are located in this region. In 13q33.3-q34 MDR, one candidate tumor suppressor known as P33ING1 has been isolated[45]. Overexpression of p33ING1 inhibited cell cycle progression and the repression function of p33ING1 was enhanced by the Ras/Raf pathway[46]. Recently, another candidate tumor suppressor known as deleted in liver cancer 2 (DLC2) was identified on 13q12.3. It shows homology to tumor suppressor gene DLC1 located at 8p22-p21.3. DLC2 encodes a protein with RhoGAP, SAM and START domains and is significantly underexpressed in only 8 of 45 (18%) HCC, as compared with its ubiquitous expression in normal tissues[47].
Loss of 16q One of the most frequent losses in HCC cases is located in the long arm of chromosome 16. Loss of 16q was detected in 30%–70% of HCC cases by CGH studies[9–13]. LOH also revealed allelic deletions in 55%–59% of HCC[14–16]. Two MDR at 16q in HCC have been reported to include 16q12.1-q23.1 (71%) and 16q23.1-q24.1 (71%)[33]. Several candidate tumor suppressors have been reported in recent publications. The loss of 16q has also been reported as a frequent genomic alteration in ovarian[27] and prostate cancer[48]. The loss of 16q24.1-q24.2 in prostate cancer has been associated with aggressive behavior of the disease, recurrent growth, poor differentiation of the tumor, and a poor prognosis for the patient[48]. The best-studied tumor suppressor gene is E-cadherin (CDH1) at 16q22.1. Inter-estingly, expression of the E-cadherin gene is reduced by a hypermethylation mechanism although no somatic mutations of the E-cadherin gene in HCC have yet been described[49,50]. Bando et al identified a distinct commonly deleted region at 16q24.1-24.2 (D16S534 and D16S3091) near novel sequence HSRG1[51]. Another candidate tumor suppressor known as WWOX was identified at 16q23.3-q24.1: absence or reduced expression of WWOX was observed in 60% of HCC cell lines compared with normal liver[52].
Loss of 17p Loss of 17p is one of the most frequent chromosomal alterations in HCC as determined by both CGH[2,11–13] and LOH[14–16]. Deletion of 17p is common in various cancers including breast[53] and colon cancer[54]. The frequent deletion of 17p may affect the tumor suppressor gene TP53 on 17p13.1. TP53 is frequently inactivated in various types of malignant tumors, including HCC[55]. However, Yumoto et al showed that loss of 17p occurred in 18/31 HCC cases (58%) by LOH, whereas TP53 mutation was only observed in 8/31 HCC cases (26%)[56]. Our recent study revealed that loss of one allele at 17p13.3 distal to the TP53 gene was observed in 48/94 HCC (51%), whereas LOH at 17p13.1, near the TP53 gene, was detected in 30/94 HCC (32%) and p53 mutation was only detected in 22/94 HCC (23%)[57]. These results suggest that another tumor suppressor gene at 17p13.3 may be involved in the pathogenesis of HCC. One candidate suppressor, C17orf25, has been isolated from this region[58]. Downregulated expression of C17orf25 was found in HCC samples matched to adjacent non-tumor liver tissues. Furthermore, transfection of C17orf25 into the hepatocellular carcinoma cell SMMC-7721 inhibited cell growth[58].
Conclusion
Molecular cytogenetic approaches such as CGH and LOH provide important clues to the identification of novel genes involved in the multistage development of HCC. In this review, we discussed some of the most common chromosome aberrations, including gains of 1q, 8q, and 20q, and loss of 4q, 8p, 13q, 16q, and 17p, followed by candidate oncogenes and tumor suppressors that may be involved within these regions. Due to the heterogeneity of HCC, it has been suggested that gains or losses of specific chromosomal loci rarely affect more than half of them, and specific HCC-related genes have not been identified in most of the above-mentioned chromosomal regions[3]. Results obtained with traditional approaches such as cytogenetic CGH and LOH studies are relatively low in resolution and are labor intensive. Advances in microarray-based approaches are beginning to reverse this situation by providing an integrated view of genetic alterations during tumorigenesis and progression[59].
Different microarray-based CGH (aCGH) platforms are emerging that allow high-resolution genomic profiling[60–62]. For instance, Hashimoto et al reported the use of cytogenetic CGH and aCGH in the identification of genes involved in HCV-related HCC[63]. In a recent article, Ishkanian et al reported the construction of submegabase resolution tiling genomic arrays (SMRT arrays) containing 32 433 overlapping BAC clones. This allows the detection of cryptic chromosomal changes down to microgain and microloss resolution[64]. In addition, some groups have used oligonucleotide arrays for high-resolution CGH and LOH studies. Single nucleotide polymorphism (SNP) arrays have been employed for high-resolution LOH studies in small cell lung cancer samples[65] and have permitted the simultaneous detection of copy number changes and LOH in a single experiment[66].
In another approach, chromosomal bias of gene expression signature was studied in HCC cases[67,68]. Using a recently developed technique known as expression balance map analysis, Midorikawa et al identified regions with frequent chromosomal aberrations in HCC based solely on expression data. Common alterations, including gain of 1q21-q23 and loss of 4q13, were found in 74% and 48% of HCC cases, respectively[67]. These matched well with previous CGH analyses. In conclusion, a combination of these powerful technologies provides a more comprehensive view of the development of HCC, and identification of novel gene candidates may provide better diagnosis and prognostication of HCC.
References
- Arbuthnot P, Kew M. Hepatitis B virus and hepatocellular carcinoma. Int J Exp Pathol 2001;82:77-100.
- Block TM, Mehta AS, Fimmel CJ, Jordan R. Molecular viral oncology of hepatocellular carcinoma. Oncogene 2003;22:5093-107.
- Thorgeirsson SS, Grisham JW. Molecular pathogenesis of human hepatocellular carcinoma. Nat Genet 2002;31:339-46.
- Miteiman F, Kaneko Y, Trent J. Human gene mapping 11: report of the committee on chromosome changes in neoplasia. Cytogenet Cell Genet 1991;58:1053-79.
- Albertson DG, Collins C, McCormick F, Gray JW. Chromosome aberrations in solid tumors. Nat Genet 2003;34:369-76.
- Yunis JJ, Ramsay N. Retinoblastoma and subband deletion of chromosome 13. Am J Dis Child 1978;132:161-3.
- Zech L, Haglund U, Nilsson K, Klein G. Characteristic chromosomal abnormalities in biopsies and lymphoid-cell lines from patients with Burkitt and non-Burkitt lymphomas. Int J Cancer 1976;17:47-56.
- Kallioniemi A, Kallioniemi OP, Sudar D, Rutovitz D, Gray JW, Waldman F, et al. Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science 1992;258:818-21.
- Marchio A, Meddeb M, Pineau P, Danglot G, Tiollais P, Bernheim A, et al. Recurrent chromosomal abnormalities in hepatocellular carcinoma detected by comparative genomic hybridization. Genes Chromosomes Cancer 1997;18:59-65.
- Wong N, Lai P, Lee SW, Fan S, Pang E, Liew CT, et al. Assessment of genetic changes in hepatocellular carcinoma by comparative genomic hybridization analysis: relationship to disease stage, tumor size, and cirrhosis. Am J Pathol 1999;154:37-43.
- Kusano N, Shiraishi K, Kubo K, Oga A, Okita K, Sasaki K. Genetic aberrations detected by comparative genomic hybridization in hepatocellular carcinomas: their relationship to clinicopathological features. Hepatology 1999;29:1858-62.
- Guan XY, Fang Y, Sham JS, Kwong DL, Zhang Y, Liang Q, et al. Recurrent chromosome alterations in hepatocellular carcinoma detected by comparative genomic hybridization. Genes Chromosomes Cancer 2000;29:110-6.
- Chang J, Kim NG, Piao Z, Park C, Park KS, Paik YK, et al. Assessment of chromosomal losses and gains in hepatocellular carcinoma assessment of chromosomal losses and gains in hepatocellular carcinoma. Cancer Lett 2002;182:193-202.
- Nishida N, Fukuda Y, Kokuryu H, Sadamoto T, Isowa G, Honda K, et al. Accumulation of allelic loss on arms of chromosomes 13q, 16q and 17p in the advanced stages of human hepatocellular carcinoma. Int J Cancer 1992;51:862-8.
- Nagai H, Pineau P, Tiollais P, Buendia MA, Dejean A. Comprehensive allelotyping of human hepatocellular carcinoma. Oncogene 1997;14:2927-33.
- Piao Z, Park C, Park JH, Kim H. Allelotype analysis of hepatocellular carcinoma. Int J Cancer. 1998;75:29-33.
- Grisham JW. Molecular genetic alterations in primary hepatocellular neoplasms: hepatocellular adenoma, hepatocellular carcinoma, and hepatoblastoma. In: Coleman WB, Tsongalis GJ, editors. The Molecular Basis of Human Cancer. Totowa, NJ: Humana Press; 2001. p 269–346.
- Wang Y, Wu MC, Sham JS, Zhang W, Wu WQ, Guan XY. Prognostic significance of c-myc and AIB1 amplification in hepatocellular carcinoma. A broad survey using high-throughput tissue microarray. Cancer 2002;95:2346-52.
- Qin LX, Tang ZY, Sham JS, Ma ZC, Ye SL, Zhou XD, et al. The association of chromosome 8p deletion and tumor metastasis in human hepatocellular carcinoma. Cancer Res 1999;59:5662-5.
- Nishimura T, Nishida N, Itoh T, Komeda T, Fukuda Y, Ikai I, et al. Discrete breakpoint mapping and shortest region of overlap of chromosome arm 1q gain and 1p loss in human hepatocellular carcinoma detected by semiquantitative microsatellite analysis. Genes Chromosomes Cancer 2004;42:34-43.
- Nishimura T, Nishida N, Itoh T, Kuno M, Minata M, Komeda T, et al. Comprehensive allelotyping of well-differentiated human hepatocellular carcinoma with semiquantitative determination of chromosomal gain or loss. Genes Chromosomes Cancer 2002;35:329-39.
- Wong N, Chan A, Lee SW, Lam E, To KF, Lai PB, et al. Positional mapping for amplified DNA sequences on 1q21-q22 in hepatocellular carcinoma indicates candidate genes over-expression. J Hepatol 2003;38:298-306.
- Sattler HP, Rohde V, Bonkhoff H, Zwergel T, Wullich B. Comparative genomic hybridization reveals DNA copy number gains to frequently occur in human prostate cancer. Prostate 1999;39:79-86.
- Tai ALS, Yan WS, Fang Y, Xie D, Sham JST, Guan XY. Recurrent chromosomal imbalances in non small cell lung cancer: the association of 1q amplification and tumor recurrence. Cancer 2004;100:1918-27.
- Kwong D, Lam A, Guan XY, Law S, Tai A, Sham J. Chromosomal aberrations in esophageal squamous cell carcinoma among Chinese: gain of 12p predicts poor prognosis after surgery. Human Pathol 2004;35:309-16.
- Fong Y, Guan XY, Guo Y, Deng M, Liang Q, Li H, et al. Analysis of genetic alterations in primary nasopharyngeal carcinoma by comparative genomic hybridization (CGH). Genes Chromosomes Cancer 2001;30:254-60.
- Sham JST, Tang TCM, Fang Y, Sun L, Qin LX, Xie D, Guan XY. Recurrent chromosome alterations in primary ovarian carcinoma of Chinese women. Cancer Genet Cytogenet 2002;133:39-44.
- Tirkkonen M, Tanner M, Karhu R, Kallioniemi A, Isola J, Kallioniemi OP. Molecular cytogenetics of primary breast cancer by CGH. Genes Chromosomes Cancer 1998;21:177-84.
- Okamoto H, Yasui K, Zhao C, Arii S, Inazawa J. PTK2 and EIF3S3 genes may be amplification targets at 8q23-q24 and are associated with large hepatocellular carcinomas. Hepatology 2003;38:1242-9.
- Guan XY, Xu J, Anzick SL, Zhang H, Trent JM, Meltzer PS. Hybrid selection of transcribed sequences from microdissected DNA: isolation of genes within amplified region at 20q11-113.2 in breast cancer. Cancer Res 1996;56:3446-50.
- Anzick SL, Kononen J, Walker RL, Azorsa DO, Tanner MM, Guan XY, et al. AIB1, a novel estrogen receptor co-activator amplified in breast and ovarian cancer. Science 1997;277:965-8.
- Piao Z, Park C, Park JH, Kim H. Deletion mapping of chromosome 4q in hepatocellular carcinoma. Int J Cancer 1998;79:356-60.
- Bando K, Nagai H, Matsumoto S, Koyama M, Kawamura N, Onda M, et al. Identification of a 1-cM region of common deletion on 4q35 associated with progression of hepatocellular carcinoma. Genes Chromosomes Cancer 1999;25:284-9.
- Jou YS, Lee CS, Chang YH, Hsiao CF, Chen CF, Chao CC, et al. Clustering of minimal deleted regions reveals distinct genetic pathways of human hepatocellular carcinoma. Cancer Res 2004;64:3030-6.
- Yeh SH, Lin MW, Lu SF, Wu DC, Tsai SF, Tsai CY, et al. Allelic loss of chromosome 4q21 approximately 23 associates with hepatitis B virus-related hepatocarcinogenesis and elevated alpha-fetoprotein. Hepatology 2004;40:847-54.
- McGowan-Jordan IJ, Speevak MD, Blakey D, Chevrette M. Suppression of tumorigenicity in human teratocarcinoma cell line PA-1 by introduction of chromosome 4. Cancer Res 1994;54:2568-72.
- Deng Q, Huang S. PRDM5 is silenced in human cancers and has growth suppressive activities. Oncogene 2004;23:4903-10.
- Liao C, Zhao M, Song H, Uchida K, Yokoyama KK, Li T. Identification of the gene for a novel liver-related putative tumor suppressor at a high-frequency loss of heterozygosity region of chromosome 8p23 in human hepatocellular carcinoma. Hepatology 2000;32:721-7.
- Yan J, Yu Y, Wang N, Chang Y, Ying H, Liu W, et al. LFIRE-1/HFREP-1, a liver-specific gene, is frequently downregulated and has growth suppressor activity in hepatocellular carcinoma. Oncogene 2004;23:1939-49.
- Yuan BZ, Miller MJ, Keck CL, Zimonjic DB, Thorgeirsson SS, Popescu NC. Cloning, characterization, and chromosomal localization of a gene frequently deleted in human liver cancer (DLC-1) homologous to rat RhoGAP. Cancer Res 1998;58:2196-9.
- Wong CM, Lee JM, Ching YP, Jin DY, Ng IO. Genetic and epigenetic alterations of DLC-1 gene in hepatocellular carcinoma. Cancer Res 2003;63:7646-51.
- Zhou X, Thorgeirsson SS, Popescu NC. Restoration of DLC-1 gene expression induces apoptosis and inhibits both cell growth and tumorigenicity in human hepatocellular carcinoma cells. Oncogene. 2004;23:1308-13.
- Fujiwara Y, Ohata H, Kuroki T, Koyama K, Tsuchiya E, Monden M, et al. Isolation of a candidate tumor suppressor gene on chromosome 8p21.3-p22 that is homologous to an extracellular domain of the PDGF receptor beta gene. Oncogene 1995;10:891-5.
- Levy A, Dang UC, Bookstein R. High-density screen of human tumor cell lines for homozygous deletions of loci on chromosome arm 8p. Genes Chromosomes Cancer 1999;24:42-7.
- Garkavtsev I, Grigorian IA, Ossovskaya VS, Chernov MV, Chumakov PM, Gudkov AV. The candidate tumour suppressor p33ING1 cooperates with p53 in cell growth control. Nature 1998;391:295-8.
- Goeman F, Thormeyer D, Abad M, Serrano M, Schmidt O, Palmero I, et al. Growth inhibition by the tumor suppressor p33ING1 in immortalized and primary cells: involvement of two silencing domains and effect of Ras. Mol Cell Biol 2005;25:422-31.
- Ching YP, Wong CM, Chan SF, Leung TH, Ng DC, Jin DY, . Deleted in liver cancer (DLC) 2 encodes a RhoGAP protein with growth suppressor function and is underexpressed in hepatocellular carcinoma. J Biol Chem 2003; 278: 10 824–30.
- Elo JP, Harkonen P, Kyllonen AP, Lukkarinen O, Vihko P. Three independently deleted regions at chromosome arm 16q in human prostate cancer: allelic loss at 16q24.1-q24.2 is associated with aggressive behaviour of the disease, recurrent growth, poor differentiation of the tumour and poor prognosis for the patient. Br J Cancer 1999;79:156-60.
- Yoshiura K, Kanai Y, Ochiai A, Shimoyama Y, Sugimura T, Hirohashi S. Silencing of the E-cadherin invasion-suppressor gene by CpG methylation in human carcinomas. Proc Natl Acad Sci USA 1995;92:7416-9.
- Wei Y, Van Nhieu JT, Prigent S, Srivatanakul P, Tiollais P, Buendia MA. Altered expression of E-cadherin in hepatocellular carcinoma: correlations with genetic alterations, beta-catenin expression, and clinical features. Hepatology 2002;36:692-701.
- Bando K, Nagai H, Matsumoto S, Koyama M, Kawamura N, Tajiri T, et al. Identification of a 1-Mb common region at 16q24.1-24.2 deleted in hepatocellular carcinoma. Genes Chromosomes Cancer 2000;28:38-44.
- Park SW, Ludes-Meyers J, Zimonjic DB, Durkin ME, Popescu NC, Aldaz CM. Frequent downregulation and loss of WWOX gene expression in human hepatocellular carcinoma. Br J Cancer 2004;91:753-9.
- Cornelis RS, van Vliet M, Vos CB, Cleton-Jansen AM, van de Vijver MJ, Peterse JL, et al. Evidence for a gene on 17p13.3, distal to TP53, as a target for allele loss in breast tumors without p53 mutations. Cancer Res 1994;54:4200-6.
- He QJ, Zeng WF, Sham JST, Xie D, Yang XW, Lin HL, et al. Recurrent genetic alterations in 26 colorectal carcinomas and 21 adenomas from Chinese patients. Cancer Genet Cytogenet 2003;144:112-8.
- Bressac B, Kew M, Wands J, Ozturk M. Selective G to T mutations of p53 gene in hepatocellular carcinoma from southern Africa. Nature 1991;350:429-31.
- Yumoto Y, Hanafusa T, Hada H, Morita T, Ooguchi S, Shinji N, et al. Loss of heterozygosity and analysis of mutation of p53 in hepatocellular carcinoma. J Gastroenterol Hepatol 1995;10:179-85.
- Guan XY, Sham JST, Tai LS, Fang Y, Li H, Liang Q. Evidence for another tumor suppressor gene at 17p13.3 distal to p53 in hepatocellular carcinoma. Cancer Genet Cytogenet 2003;140:45-8.
- Qin WX, Wan F, Sun FY, Zhang PP, Han LW, Huang Y, et al. Cloning and characterization of a novel gene (C17orf25) from the deletion region on chromosome 17p13.3 in hepatocellular carcinoma. Cell Res 2001;11:209-16.
- Pollack JR, Iyer VR. Characterizing the physical genome. Nat Genet 2002;32:515-21.
- Pinkel D, Segraves R, Sudar D, Clark S, Poole I, Kowbel D, et al. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat Genet 1998;20:207-11.
- Albertson DG, Pinkel D. Genomic microarrays in human genetic disease and cancer. Hum Mol Genet 2003;12:R145-52.
- Mantripragada KK, Buckley PG, de Stahl TD, Dumanski JP. Genomic microarrays in the spotlight. Trends Genet 2004;20:87-94.
- Hashimoto K, Mori N, Tamesa T, Okada T, Kawauchi S, Oga A, et al. Analysis of DNA copy number aberrations in hepatitis C virus-associated hepatocellular carcinomas by conventional CGH and array CGH. Mod Pathol 2004;17:617-22.
- Ishkanian AS, Malloff CA, Watson SK, DeLeeuw RJ, Chi B, Coe BP, et al. A tiling resolution DNA microarray with complete coverage of the human genome. Nat Genet 2004;36:299-303.
- Lindblad-Toh K, Tanenbaum DM, Daly MJ, Winchester E, Lui WO, Villapakkam A, et al. Loss-of-heterozygosity analysis of small-cell lung carcinomas using single-nucleotide polymorphism arrays. Nat Biotechnol 2000;18:1001-5.
- Zhao X, Li C, Paez JG, Chin K, Janne PA, Chen TH, et al. An integrated view of copy number and allelic alterations in the cancer genome using single nucleotide polymorphism arrays. Cancer Res 2004;64:3060-71.
- Midorikawa Y, Tsutsumi S, Nishimura K, Kamimura N, Kano M, Sakamoto H, et al. Distinct chromosomal bias of gene expression signatures in the progression of hepatocellular carcinoma. Cancer Res 2004;64:7263-70.
- Patil MA, Chua MS, Pan KH, Lin R, Lih CJ, Cheung ST, et al. An integrated data analysis approach to characterize genes highly expressed in hepatocellular carcinoma. Oncogene 2005;21:1-11.