
Antibody engineering: facing new challenges in cancer therapy1
Introduction
Monoclonal antibodies (mAb) are unique and versatile molecules that have been found applications in research, diagnosis, and in the treatment of multiple diseases, including cancer. The advent of hybridoma technology for mAb production in 1975[1] was a breakthrough in the field of biomedicine; 30 years later, a plethora of biotech companies produces thousands of mAb, and at least 17 of them have FDA (US Food and Drug Administration) approval for therapeutic use in patients, with hundreds of them still in the pipeline.
However promising their future is, the development of therapeutic mAb suffered a number of serious drawbacks, which considerably reduced faith in their clinical applicability. These disappointments were caused by their inability to trigger human effector functions, and because repeated administration provoked an immune response against murine antibody (Ab) domains (HAMA, human anti-murine Abs)[2]. Recently developed technologies (phage display and transgenic mice) allow the selection and identification of fully human Ab, as well as the improvement of Ab affinity[3]. The ability to generate human mAb achieved 2 important goals: it overcame most host anti-Ab responses, and it extended the half-life of the reagent to something closer to that of normal IgG. As a result of these advances, mAb are starting to fulfill their potential as therapeutics.
Not surprisingly, Ab engineering has constituted its own field. Mutations can be introduced in the variable regions to increase the affinity of the Ab for its antigen or in the constant region to enhance its natural effector functions. Pharmacokinetics and avidity are improved by multimerization of Ab fragments. Ab molecules have been dissected to their basic elements, and then rearranged to produce a variety of formats not found in nature that display new properties. Moreover, these “building blocks” have been incorporated into multiple types of fusion proteins, soluble (immunocytokines, immunotoxins), as well as part of artificial cell surface receptors and viral envelopes for the retargeting of both effector cells and virus particles (Figure 1).
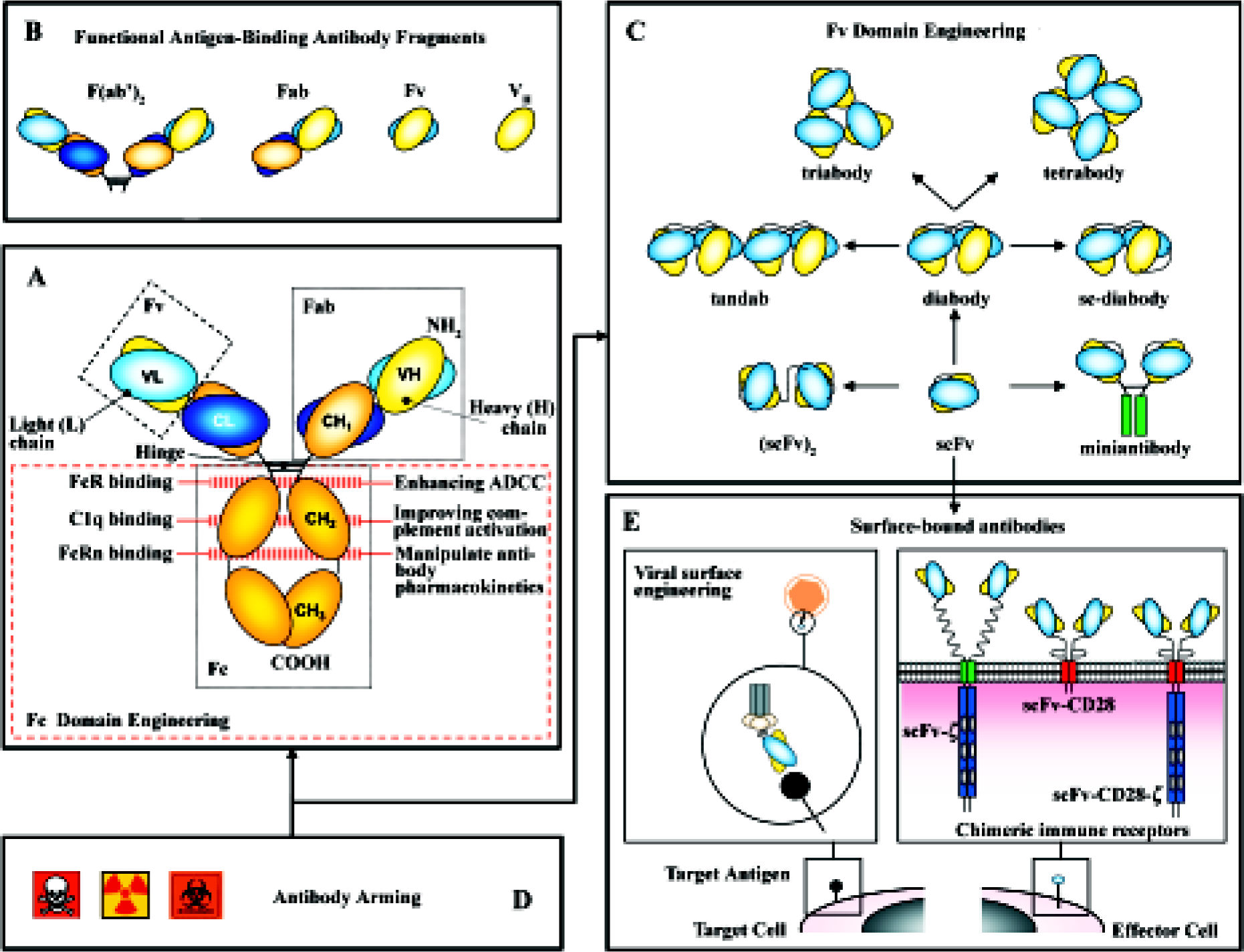
Making better mAb: how to improve their “natural” effector functions
Ab contains two functionally and molecularly separable modules (Figure 1A): one module for antigen binding (Fab) and another for triggering effector functions (Fc). The antigen-binding region can be manipulated to increase both binding affinity and specificity. Methods of Ab affinity maturation are based on the principle of changing parts of the variable domains while keeping the specificity. Different approaches are: chain-shuffling (substitution of the native light chain with a new light-chain repertoire, but retaining the variable heavy chain), randomization of complementarity determining regions (CDR), and generation of Ab libraries with mutations within the variable regions by error-prone PCR, and E coli mutator strains or site-specific mutagenesis[4].
Fragment cystalizable (FC) domains contain motifs for the activation of both effector immune cells and the classical pathway of the complement (C1q) responsible, respectively, for Ab-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC). A variety of immune cells express on their surface receptors for the Fc domain of IgG1 and IgG3: FcγRI, FcγRIIa, FcγRIII are stimulatory and FcγRIIb is inhibitory. The Fc domain sequence can be manipulated (Figure 1A) to increase its affinity for the stimulatory receptors or to decrease its binding to the inhibitory one, modulating the activity of the whole Ab[5]. Similarly, point mutations in the Fc domain can increase the binding to C1q, improving complement activation[6]. Modification of Fc glycosylation can also enhance its ability to support ADCC[7].
The Fc region of human Ab also contains a binding motif for the receptor FcRn that protects immunoglobulins (Ig) from intracellular degradation (Figure 1A). By contrast, rodent mAb fail to bind to FcRn and are rapidly removed from the circulation. Using combinatorial phage display libraries, mutations in the Fc region have been identified with higher binding affinity to FcRn, implying longer half-life of the mAb[8].
New formats, new functions
The domain architecture of Ig has facilitated the creation of both smaller and larger forms with variable valency for one or more target antigens and pharmacokinetic properties that are tunable to specific settings[9]. Non-natural Ab formats (Figure 1C), such as the single-chain fragment variable (scFv) and the diabody, are rapidly emerging as key players in the engineered Ab field. A scFv (~30 kDa) comprises the V domains of the heavy and light chains (VH and VL) of a mAb joined by a linker sequence[3]. A diabody (~60) kDa is produced when scFv contain short interdomain linkers (5 aminoacids or less)[10]. This prevents intramolecular pairing of the VH and VL domains on the same chain, but allows interchain pairing to form dimers. Reducing further the linker length promotes the assembly of scFv into trimers (tria-bodies, ~90 kDa) or tetramers (tetrabodies, ~120 kDa). The increased binding valency of these multimers results in high avidity and low off-rates.
Given that Ab fragments lack the Fc region, their biological effects can not be attributed to CDC or ADCC. In this context, the nature of the target is crucial, as Ab fragments function by blocking the action of specific molecules or by acting as signaling molecules. The blocking activity is achieved by preventing growth factors, cytokines or other soluble mediators reaching their target receptors, accomplished either by the Ab binding to the factor itself or to its receptor. The signaling effect is based on the crosslinking of receptors that are, in turn, connected to mediators of cell division or programmed cell death[4].
Diabodies constitute the most effective way to generate bispecific Ab fragments through their ability to bind to 2 different antigens and used to crosslink various cells and molecules. Bispecific Ab offer a variety of new effector mechanisms: retargeting of effector cells (cytotoxic T cells, NK cells, and macrophages), recruitment of effector molecules (toxins, drugs, prodrugs, cytokines, radioisotopes, and complement system) and retargeting of carrier systems (viral vectors for gene therapy)[11].
Ab fragments as Fab and scFv offer several advantages because of their small size when compared to parental Ig (150 kDa): (i) they are easy to produce in bacterial systems; (ii) extravasate more efficiently; and (iii) their tissue penetration ability is higher. However, recombinant proteins that are smaller than 60 kDa are taken up by the kidney and excreted into the urine. Therefore, these molecules tend to have a short circulating half-life. As Fab and scFv fragments lack the Fc region altogether, they can not be saved from degradation by FcRn. Multimerization is an obvious strategy to increase the size, and therefore, the half-life of Ab fragments (triabodies, tetrabodies). The use of bispecific Ab fragments for retargeting serum Ig provides with the Fc-associated effector functions and prolongs the residence time in serum. A new approach is the pegylation of Ab fragments, achieved by chemical coupling of polyethylene glycol (PEG) to amino groups in the protein structure, increasing the size of the molecule above the glomerular filtration limit[12].
Naked versus armed mAb: acquisition of new effector functions
In cancer therapy, Ab fragments are fused (chemically or at genetic level) with a range of molecules to introduce different functionalities, including cytotoxic drugs, toxins, or radionuclides for cancer cell killing, enzymes for prodrug therapy and cytokines to stimulate the antitumor immune response (Figure 1D). These “armed” Ab exhibit considerably better therapeutic performance than their “naked” counterparts (for a review on the topic, see Ref [2]).
Incorporation of agents with direct toxic effect The most widely explored strategy for enhancing the efficacy of antitumor Ab is direct arming by linkage to cytotoxic agents or radionuclides. In fact, 3 of the approved mAb for use in patients belong to this group: gemtuzumab ozogamicin was the first (2000), followed by ibritumomab tiuxetan and tositumomab (2002 and 2003, respectively). Gemtuzumab ozogamicin is an anti-CD33 (antigen expressed in 90% of acute myeloid leukemias) mAb conjugated to calicheamicin. Calicheamicins and maytansinoids are (100–1000)-fold more potent than conventional chemotherapeutics and constitute the most extensively evaluated small-molecule toxins used for Ab arming[2]. Biological toxins, such as ricin or diphtheria toxin, can be attached to an Ab (native Ig or recombinant fragments), although their clinical application has been hampered by their high toxicity. It has recently been published that a single point mutation in ricin toxin can eliminate vascular damage without compromising its action[13].
Ibritumomab tiuxetan and tositumomab are anti-CD20 mAb conjugated, respectively, to 90Y and 131I, and approved for non-Hodgkin lymphoma treatment. Radioimmunotherapy (RAIT/RIT) has the advantage to kill bystander cells, especially interesting when not all the tumor cells express the antigen recognized by the Ab[14].
ADEPT: Ab-directed enzyme prodrug therapy ADEPT involves the pre-targeting of prodrugs to tumors. An Ab-enzyme fusion protein is first administered and allowed to localize to the tumor, followed by the administration of the prodrug which is activated by the enzyme at the tumor site[15,16]. This strategy has proven highly effective in preclinical tumor models, allowing 4–12 fold higher intratumor drug concentrations and up to 5-fold lower extratumor drug concentration[2]. An interesting approach is the use of Abs with inherent catalytic activity, so the conjugation to an enzyme is not required[17].
Immunocytokines Several cytokines have demonstrated their potent antitumoral effect, but unfortunately their side effects limit their administration. In order to accumulate preferentially the cytokine in the tumor, fusion proteins (immunocytokines) consisting of a targeting mAb specific for a tumor antigen and the selected cytokine (IL-2, IL-12, TNF-α and GM-CSF) have been designed[18]. These immunocytokines allow the local activation of the antitumoral immune response, avoiding the toxicity associated with systemic cytokine administration[19,20]. A different approach is based on the targeting of tumor vasculature using cyto-kines with recognized antiangiogenic effect, as IL-12[21].
Gene therapy: new scenarios
The practical utility of Ab fragments has been limited by problems related to large-scale production and biodistri-bution. Monovalent Ab fragments exhibit rapid blood clearance and poor retention time on the target, which results in the necessity of frequent delivery of such Ab fragments. To circumvent these limitations, Ab-based gene therapy approaches have been developed. In vivo production makes the Abs less immunogenic and better tolerated and results in effective and persistent levels of Ab fragments, compensating for the rapid blood clearance of scFvs. Moreover, genetic approaches provide Ab molecules with new functions in unexpected scenarios[22].
Secretion of soluble Abs by genetically modified cells In vivo production of therapeutic mAb by genetically engineered cells could advantageously replace the injection of purified Ab in cancer treatment. The feasibility of the in vivo production and systemic delivery of mAb by different cells/tissues has now been demonstrated using different techni-ques, as ex vivo genetically modified autologous or encapsulated heterologous cells and in vivo gene transfer using viral vectors[23].
In the first work reporting a therapeutic effect associated to in vivo mAb production, an anti-erbB-2 scFv was expressed using an adenoviral vector. In this model, a human ovarian cancer cell line erbB-2+ was established in the context of athymic nude mice. Whereas exponential growth in tumor volumes was noted in the control groups, a clear inhibition of tumor growth was observable for the animals treated with the adenoviral vector encoding anti-erbB-2 scFv[24].
We have demonstrated that both monospecific and bispecific Ab can be efficiently produced by mammalian cells with a clear therapeutic effect. Using an anti-laminin scFv with antiangiogenic activity[25,26], we assessed that gene-modified human fibrosarcoma cells failed to grow to detectable tumors when inoculated in athymic mice[27]. In another set of experiments, functionally active diabody (anti-CEA x anti-CD3) was secreted from stably transfected human cells and promoted unstimulated human primary T cells to proliferate and kill CEA-expressing cancer cells. Importantly, locally produced diabodies showed significant cytotoxic activity in vivo against established tumors and only required the infusion of small numbers of functional T cells[28].
Surface-bound Abs: chimeric immune receptors Adoptive cellular immunotherapy of cancer has been limited mostly because of the poor immunogenicity of tumor cells and the difficulties in obtaining tumor-specific MHC-restricted cytotoxic T lymphocytes (CTL) in large numbers[29]. To circumvent these limitations, new strategies have been designed in order to target CTL to relevant tumor cell surface antigens, including genetic manipulation of T cells to graft them with new recognition specificities[30].
Chimeric immune receptors (CIR) genes are composed of a recognition unit attached to the transmembrane and intracytoplasmic sequences of a signaling molecule. Most Ab-derived CIR use scFvs as recognition domains. Signaling molecules belong to a family of structurally and functionally related proteins that include TCR-associated polypeptides and some Fc receptors. As the requirements of MHC restriction are bypassed, the tumor cell recognition of CTL grafted with CIR is not hampered by the down-regulation of HLA class I molecules usually found in tumors[30].
The utility and effectiveness of the CIR approach has been demonstrated in a variety of animal models where tumor-specific CIR drove the adoptive transferred autologous T-lymphocytes to accumulate at the tumor site in vivo and prevented the growth of syngenic tumors that grow rapidly in the native host. Target antigens include CEA (colorectal cancer), PSMA (prostate cancer), erbB-2 (breast and others), CD19 and CD20 (B-cell malignancies), CD30 (lymphomas), GD2 (neuroblastoma) and the tumor neovasculature receptor VEGFR-2[22]. Recently, human peripheral blood leukocytes genetically modified to target CD19 were shown to eliminate systemic B-cell tumors in immunodeficient mice[31]. Primarily investigated in T cells, CIR have also proven useful in the retargeting of NK cells[32].
Recent studies have established that the provision of additional or co-stimulatory signals is essential for the expansion and activity of adoptively transferred T cells. We have reported that CD28-based CIRs were stably expressed as functional cell surface receptors and that Ag-specific co-stimulatory signals could synergize with signals mediated through the native TCR/CD3 complex or TCRz-based CIR to produce optimal levels of IL-2[33]. Moreover, CIR providing both primary and costimulatory signaling in T cells from a single gene product have been described[34].
Viral surface engineering Most of the viral vectors developed for gene therapy have a broad tissue tropism. The development of viral vectors targeted into a selected type of cell or tissue, without losing virus infection efficiency or causing toxicity, is critical for their clinic application. Cancer cells represent major targets in this strategy, as they often express lower levels of viral receptors compared to normal cells[35].
Using Ab directed against tumor-associated antigens expressed on the cell surface for virus pseudotyping has been successfully associated with different viral vectors in directing them to cancer cells. Adenovirus (AdV) has been extensively studied in this approach, especially in the context of capsid engineering. The most recent developments in this approach have overcome several limitations in this strategy, including the need for correct ligand folding, the structural and biosynthetic compatibility of ligands with the AdV, along with the fact that in this strategy, viruses have to be specifically engineered for each particular targeting situation. Thus, several groups have used similar strategies by incorporating an IgG-binding domain of staphylococcal protein A into the AdV fiber protein, allowing the vectors to form a stable complex with either full size mAb or fusion proteins consisting of a targeting scFv fused to an Ig Fc domain[36,37].
Another group of preferred vectors in preclinical and clinical settings for cancer gene therapy are the murine leukemia virus (MLV)-based retroviral vectors. Several groups have inserted a scFv moiety in the virus envelop to obtain cell-specificity[38–41]. Using a different strategy, the IgG-binding domain of protein A was inserted into the envelope, allowing the redirecting lentiviral vectors to target cells through adaptors as described for AdV[42].
The attenuated measles virus (MV) is another vector with great therapeutic potential in gene therapy. The feasibility to expand MV tropism by virtue of a scFv displayed on its H protein was demonstrated using an anti-CEA scFv[43]. Replicating MV have been obtained which are capable of entering CD20+ or CD38+ target cells through interaction between either an anti-CD20 or anti-CD38 scFv and the cognate antigen molecules on the cell surface. Both studies have shown significant antitumor effects in vivo[44,45].
Intrabodies Intracellular Ab (intrabodies) constitute neutralizing molecules with a great potential in gene therapy and represent an alternative to other methods of gene inactivation as antisense RNA and RNA interference (RNAi). When provided with the corresponding protein trafficking signals, intrabodies can be directed to endoplasmic reticulum via addition of a SEKDEL retention signal, nucleus via the SV40-derived nuclear localization signal, inner face of the plasma membrane by the addition of farnesylation signals or cytoplasm simply by the deletion of the leader peptide. Although classically designed to divert proteins from their usual cellular compartment or to block protein-protein or protein-nucleic acid interactions, this concept is currently in expansion, with intrabodies capable of directly inhibiting the function of an enzyme, activating intracellular proteins, as caspase-3, or leading proteins to degradation in the ubiquitin-proteasome pathway using F-box-intrabody fusions[46].
ScFvs are the preferred format for intrabodies, but their stability is affected by the reducing conditions inside the cell, which prevent the formation of intradomain disulphide bonds and blocks their proper folding. Efforts to generate functional intrabodies include the isolation of naturally occurring intrabodies from large libraries and the creation of an artificial intrabody framework that relies on the pre-determined ability of certain scFv to fold adequately and remain stable in the cellular milieu[46,47]. While scFv intrabodies are the most common, alternative formats have been shown to be equally effective, including single Ab domains (“came-lized” or not) and bispecific Ab, known as “intra-diabodies” which are able to downregulate simultaneously 2 cell surface receptors[48].
Regarding their application in cancer therapy, intrabodies are suitable to downregulate proteins overexpressed in tumors, such as EGFR, erbB-2, cathepsin L[49], and cyclin E or to target mutant oncogenic forms of Ras and p53 and fusion proteins as BCR-ABL. Apoptosis of tumor cells can be promoted by downregulation of Bcl-2 or activation of caspase-3, and the uptake of cytotoxic drugs can be increased blocking the multidrug resistance (MDR) gene product[22]. Inhibition of tumor neovascularization is a promising approach for cancer therapy. Recently, an adenoviral vector was used to deliver a scFv capable of blocking surface expres-sion of an endothelial cell-specific receptor and significantly inhibited growth of human xenografts in a murine model[50].
Conclusions
Antibody engineering represents an emerging technology that holds great promise for medical science. With the plethora of new molecular techniques at hand, many innovative approaches to diagnostics and therapeutics applications are under consideration. The development of protein engineering techniques to reduce immunogenicity, alter half-life, improve efficacy, and increase tumor targeting has provided the new types of antibodies that are moving rapidly from the bench to the clinic. In fact, engineered antibodies now represent over 30% of biopharmaceuticals in clinical trials. Furthermore, genetic approaches provide antibody molecules with new functions in unexpected scenarios: expression of antibody domains in precise intracellular locations and grafting of new binding activities to engineered cells. Further improvement will require the design of in vivo selection systems to generate antibodies fully active in specific cellular compartments, and the use of antibodies as tools for functional gene identification and drug target validation (genomics- and proteomics-based high-throughput systems) and for better understanding of disease pathways.
References
- Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975;256:495-7.
- Carter P. Improving the efficacy of antibody-based cancer therapies. Nat Rev Cancer 2001;1:118-29.
- Hudson PJ, Souriau C. Engineered antibodies. Nat Med 2003;9:129-34.
- Brekke OH, Sandlie I. Therapeutic antibodies for human diseases at the dawn of the twenty-first century. Nat Rev Drug Disc 2003;2:52-62.
- Shields RL, Namenuk AK, Hong K, Meng YG, Rae J, Briggs J, et al. High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J Biol Chem 2001;276:6591-604.
- Idusogie EE, Wong PY, Presta LG, Gazzano-Santoro H, Totpal K, Ultsch M, et al. Engineered antibodies with increased activity to recruit complement. J Immunol 2001;166:2571-5.
- Umaña P, Jean-Mairet J, Moudry R, Amstutz H, Bailey JE. Engineered glycoforms of an antineuro-blastoma IgG 1 with optimized antibody-dependent cellular cytotoxic activity. Nat Biotechnol 1999;17:176-80.
- Ghetie V, Popov S, Borvak J, Radu C, Matesoi D, Medesan C, et al. Increasing the serum persistence of an IgG fragment by random mutagenesis. Nat Biotechnol 1997;15:637-40.
- Pluckthun A. Mono- and bivalent antibody fragments produced in Escherichia coli: engineering, folding and antigen binding. Immunol Rev 1992;130:151-88.
- Holliger P, Prospero T, Winter G. “Diabodies”: small bivalent and bispecific antibody fragments. Proc Natl Acad Sci USA 1993;90:6444-8.
- Kontermann RE. Recombinant bispecific antibodies for cancer therapy. Acta Pharmacol Sin 2005;26:1-9.
- Natarajan A, Xiong CY, Albrecht H, DeNardo GL, DeNardo SJ. Characterization of site-specific ScFv PEGylation for tumor-targeting pharmaceuticals. Bioconjug Chem 2005;16:113-21.
- Smallshaw JE, Ghetie V, Rizo J, Fulmer JR, Trahan LL, Ghetie MA, et al. Genetic engineering of an immunotoxin to eliminate pulmonary vascular leak in mice. Nat Biotechnol 2003;21:387-91.
- Goldenberg DM. Advancing role of radiolabeled antibodies in the therapy of cancer. Cancer Immunol Immunother 2003;52:281-96.
- Xu G, McLeod HL. Strategies for enzyme/prodrug cancer therapy. Clin Cancer Res 2001;7:3314-24.
- Sharma SK, Pedley RB, Bhatia J, Boxer GM, El-Emir E, Qureshi U, et al. Sustained tumor regression of human colorectal cancer xenografts using a multifunctional mannosylated fusion protein in antibody-directed enzyme prodrug therapy. Clin Cancer Res 2005;11:814-25.
- Shabat D, Lode HN, Pertl U, Reisfeld RA, Rader C, Lerner RA, et al. In vivo activity in a catalytic antibody-prodrug system: antibody catalyzed etoposide prodrug activation for selective chemotherapy. Proc Natl Acad Sci USA 2001;98:7528-33.
- Nissim A, Gofur Y, Vessillier S, Adams G, Chernajovsky Y. Methods for targeting biologicals to specific disease sites. Trends Mol Med 2004;10:269-74.
- Hombach A, Heuser C, Abken H. Simultaneous targeting of IL2 and IL12 to Hodgkin’s lymphoma cells enhances activation of resting NK cells and tumor cell lysis. Int J Cancer. 2005;115:241-7.
- Gillies SD, Lan Y, Williams S, Carr F, Forman S, Raubitschek A, et al. An anti-CD20-IL-2 immunocytokine is highly efficacious in a SCID mouse model of established human B lymphoma. Blood. 2005;105:3972-8.
- Halin C, Rondini S, Nilsson F, Berndt A, Kosmehl H, Zardi L, et al. Enhancement of the antitumor activity of interleukin-12 by targeted delivery to neovasculature. Nat Biotechnol 2002;20:264-9.
- Sanz L, Blanco B, Alvarez-Vallina L. Antibodies and gene therapy: teaching old ‘magic bullets’ new tricks. Trends Immunol 2004;25:85-91.
- Pelegrin M, Gros L, Dreja H, Piechaczyk M. Monoclonal antibody-based genetic immunotherapy. Curr Gene Ther 2004;4:347-56.
- Arafat WO, Gomez-Navarro J, Buchsbau DJ, Xiang J, Wang M. Effective single chain antibody (scFv) concentrations in vivo via adenoviral vector mediated expression of secretory scFv. Gene Ther 2002;9:256-62.
- Sanz L, Kristensen P, Russell SJ, Ramirez Garcia JR, Alvarez-Vallina L. Generation and characterization of recombinant human antibodies specific for native laminin epitopes: potential application in cancer therapy. Cancer Immunol Immunother 2001;50:557-65.
- Sanz L, Garcia-Bermejo L, Blanco FJ, Kristensen P, Feijoo M, Suarez E, et al. A novel cell binding site in the coiled-coil domain of laminin involved in capillary morphogenesis. EMBO J 2003;22:1508-17.
- Sanz L, Kristensen P, Blanco B, Facteau S, Russell SJ, Winter G, et al. Single-chain antibody-based gene therapy: inhibition of tumor growth by in situ production of phage-derived human antibody fragments blocking functionally active sites of cell-associated matrices. Gene Ther 2002;9:1049-53.
- Blanco B, Holliger P, Vile R, Alvarez-Vallina L. Induction of human T lymphocyte cytotoxicity and inhibition of tumor growth by tumor-specific diabody-based molecules secreted from gene-modified bystander cells. J Immunol 2003;171:1070-7.
- Baxenavis CN, Papamichail M. Targeting of tumor cells by lymphocytes engineered to express chimeric receptor genes. Cancer Immunol Immunother 2004;53:893-903.
- Álvarez-Vallina L. Genetic approaches for antigen-selective cell therapy. Curr Gene Ther 2001;4:385-97.
- Brentjens RJ, Latouche JB, Santos E, Marti F, Gong MC, Lyddane C, et al. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat Med 2003;9:279-86.
- Uherek C, Tonn T, Uherek B, Becker S, Schnierle B, Klingemann HG, et al. Retargeting of natural killer-cell cytolytic activity to ErbB2-expressing cancer cells results in efficient and selective tumor cell destruction. Blood 2002;100:1265-73.
- Alvarez-Vallina L, Hawkins RE. Antigen-specific targeting of CD28-mediated T cell co-stimulation using chimeric single-chain antibody variable fragment-CD28 receptors. Eur J Immunol 1996;26:2304-9.
- Finney HM, Lawson AD, Bebbington CR, Weir AN. Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J Immunol 1998;161:2791-7.
- Sanz L, Qiao J, Vile R, Alvarez-Vallina L. Antibody engineering, virus retargeting and cellular immunotherapy: one ring to rule them all? Curr Gene Ther 2005;5:63-70.
- Korokhov N, Mikheeva G, Krendelshchikov A, Belousova N, Simonenko V, Krendelshchikov V, et al. Targeting of adenovirus via genetic modification of the viral capsid combined with a protein bridge. J Virol 2003;77:12931-40.
- Volpers C, Thirion C, Biermann V, Hussmann S, Kewes H, Dunant P, et al. Antibody-mediated targeting of an adenovirus vector modified to contain a synthetic immunoglobulin g-binding domain in the capsid. J Virol 2003;77:2093-104.
- Russell SJ, Hawkins RE, Winter G. Retroviral vectors displaying functional antibody fragments. Nucleic Acids Res 1993;21:1081-5.
- Somia NV, Zoppe M, Verma IM. Generation of targeted retroviral vectors by using single-chain variable fragment: an approach to in vivo gene delivery. Proc Natl Acad Sci USA 1995;92:7570-4.
- Chu TH, Dornburg R. Toward highly efficient cell-type-specific gene transfer with retroviral vectors displaying single-chain antibodies. J Virol 1997;71:720-5.
- Martin F, Neil S, Kupsch J, Maurice M, Cosset F, Collins M. Retrovirus targeting by tropism restriction to melanoma cells. J Virol 1999;73:6923-9.
- Morizono K, Xie Y, Ringpis GE, Johnson M, Nassanian H, Lee B, et al. Lentiviral vector retargeting to P-glycoprotein on metastatic melanoma through intravenous injection. Nat Med 2005;11:346-52.
- Hammond AL, Plemper RK, Zhang J, Schneider U, Russell SJ, Cattaneo R. Single-chain antibody displayed on a recombinant measles virus confers entry through the tumor-associated carcinoembryonic antigen. J Virol 2001;75:2087-96.
- Bucheit AD, Kumar S, Grote DM, Lin Y, von Messling V, Cattaneo RB, et al. An oncolytic measles virus engineered to enter cells through the CD20 antigen. Mol Ther 2003;7:62-72.
- Peng KW, Donovan KA, Schneider U, Cattaneo R, Lust JA, Russell SJ. Oncolytic measles viruses displaying a single-chain antibody against CD38, a myeloma cell marker. Blood 2003;101:2557-62.
- Lobato MN, Rabbits TH. Intracellular antibodies and challenges facing their use as therapeutic agents. Trends Mol Med 2003;9:390-6.
- Stocks MR. Intrabodies: production and promise. Drug Discov Today 2004;9:960-6.
- Jendreyko N, Popkov M, Beerli RR, Chung J, McGavern DB, Rader C, et al. Intra-diabodies: bispecific, tetravalent antibodies for the simultaneous functional knockout of two cell surface receptors. J Biol Chem 2003;278:47812-9.
- Rousselet N, Mills L, Jean D, Tellez C, Bar-Eli M, Frade R. Inhibition of tumorigenicity and metastasis of human melanoma cells by anti-cathepsin L single chain variable fragment. Cancer Res 2004;64:146-51.
- Popkov M, Jendreyko N, McGavern DB, Rader C, Barbas CF III. Targeting tumor angiogenesis with adenovirus-delivered anti-Tie-2 intrabody. Cancer Res 2005;65:972-81.