
Rhodanine derivatives as novel peroxisome proliferator-activated receptor γ agonists1
Introduction
Peroxisome proliferator-activated receptor (PPAR) γ, one of the subtypes of PPARs, is a ligand-dependent transcription factor of the nuclear hormone receptor superfamily[1]. PPARγ is mainly present in the adipose tissue, skeletal muscle, and liver[2]. Two other family members include PPARα, which is present in the liver, kidney and heart, and the ubiquitously-expressed PPARδ[3]. Upon ligand binding, PPARs release relevant corepressors and form heterodimers with retinoid X receptors (RXRs)[4]. The heterodimers bind to peroxisome proliferator response elements (PPREs) and recruit co-activa-tors to initiate the transcription of target genes[5,6].
Type 2 diabetes mellitus is a heterogeneous disease resulting from a dynamic interaction between defects in insulin secretion and insulin action[7]. In animal models and humans affected by this disorder, treatment with thiazo-lidinediones decreases elevated plasma glucose, triglyceride, and insulin concentrations[8,9]. Their actions also lead to the stimulation of adipogenesis and the recovery of insulin sensitivity in the target tissues[9]. The antidiabetic effects of thiazolidinediones are directly mediated through binding to PPARγ, thereby resulting in an active conformation of the receptor[10].
While the clinical benefits of PPARγ agonists in treating type 2 diabetes have been clearly demonstrated, the current generation of glitazones is associated with undesired side-effects, such as weight gain and edema[11]. Thus, there has been significant interest in the design of novel PPARγ-modulating drugs that retain efficacious insulin sensitizing properties while minimizing potential adverse effects, especially for long-term use.
SH00012671 is a core structure discovered in a high-throughput screening campaign directed towards PPARγ[12]. It displayed a relatively higher binding affinity to the receptor, but was almost inactive in cell-based assays. In this paper, we describe the design, synthesis, and biological evaluation of a series of analogues to SH00012671 with the aim of identifying novel PPARγ agonists.
Materials and methods
Reagents Edetic acid, insulin, and dexamethasone were purchased from Sigma-Aldrich (St Louis, MO, USA). BRL49653 (rosiglitazone) was bought from Cayman Chemical Co (Ann Arbor, MI, USA). [3H]BRL49653 (53 Ci/mmol) was obtained from American Radiolabeled Chemicals (St Louis, MO, USA), FlashPlate and flat-bottom Isoplate were from PerkinElmer (Boston, MA, USA), and the streptavidin-coated microbeads were from Amersham Biosciences UK (Buckinghamshire, England). The plasmids of human nuclear receptors used in this study were from Dr Xu SHEN of Shanghai Institute of Materia Medica, Chinese Academy of Sciences (Shanghai, China) and Dr Sai-juan CHEN of Shanghai Institute of Hematology (Shanghai, China). Full-length PPARγ and RXRα were produced with a baculovirus expression system using fall armyworm (Spodoptera frugiperda) immature ovarian (IPLB-SF-21-AE) cells[13]. The PPRE reporter vector was purchased from Panomics (Redwood City, CA, USA), the Steady-Glo luciferase assay system was from Promega (Madison, WI, USA), the FuGene 6 transfection reagent was from Roche Diagnostics (Indianapolis, IN, USA), and the triglyceride (TG) detection kit was from Ningbo Asia-Pacific Biotechnology Co (Zhejiang, China).
Chemistry Figure 1 depicts the syntheses of the rho-danine derivatives designed based on SH00012671. Esterification of aromatic acid 1 by methanol and then hydrazinolysis of the resulting methyl ester provided the substituted benzoyl hydrazine 2. The reaction of 2 with carbon disulphide in ethanol in the presence of potassium hydroxide for 2 h resulted in the formation of potassium β-acyldithiocarbazinate 3, which was used without further purification. According to Tadashi and Masaki[14] when 3 was added to a solution of chloroacetic acid and saturated sodium carbonate, stirred at room temperature for 30 min, and acidified by aquous HCl, 3-benzoylamino rhodanine 4 would be separated out and then solidify. However, we found that when acidifying with aquous HCl, the intermediate (2-benzoyl-thiocarbazyl) mercaptoacetic acid, instead of 3-benzoyl-amino rhodanine 4, was separated out as evidenced by 1H-NMR (Nuclear magnetic resonance) and ESI (Electrospray ioniza-tion). This intermediate was thus refluxed in dioxane for 1 h to give 3-benzoylaminorhodanine 4, which could also be obtained by dissolving 3-amino rhodanine 5 in pyridine with benzoyl chloride. The final compounds 7–19 were obtained through the condensation of 4 with benzoaldehyde 6 by refluxing in glacial acetic acid in the presence of sodium acetate.
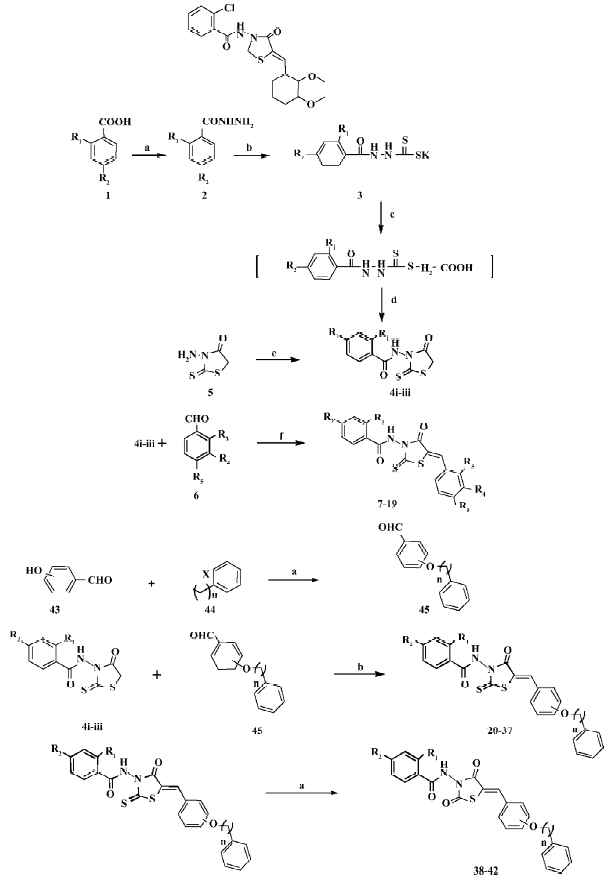
Next, we changed the substitutions on R4 or R5 while R1 or R2 was fixed with chloro (Cl), Br (bromo), or H (hydrogen; Figure 1). We used p-hydroxy benzaldehyde or m-hydroxy benzaldehyde 43 as the raw material. It was alkylated with different halogenated aralkyl 44 of which the chain length was changed from 1 to 3. Compounds 20–37 were obtained through the condensation of 4 with benzoaldehyde 45 by refluxing in glacial acetic acid in the presence of sodium acetate. Compounds 38–42 were made by substituting thio on the rhodanine with oxygen, in which the former rhodanine was refluxed with excessive chloroacetic acid in ethanol and water (Figure 1).
Receptor-binding assay Biotinylated PPRE (2 µL from a stock solution of 10 g/L) was mixed with the assay buffer (10 mL) containing fish sperm DNA (Shanghai Sangon Biotechnology Co, Shanghai, China; 10 µL from a 10 g/L stock solution) and 4 mg streptavidin-coated microbeads in a conical polypropylene centrifuge tube (Corning, Corning, NY, USA) and incubated overnight at 4 °C. The mixture was centrifuged for 10 min at 1 500×g. The supernatant was then removed and washed 3 times with 10 mL of the assay buffer. The reaction solution 10 mL containing 700 µg human PPARγ extract protein (70 mg/L), 47 µg human RXRα extract protein (4.7 mg/L), 10 nmol/L [3H]BRL49653, and various concentrations of BRL49653 or test compounds were distributed to each well of the Isoplate (100 µL/well) and incubated at 4°C for 4 h before counting by the MicroBeta counte (PerkinElmer, USA).
Cotransfection assay The CV-1 cells (1×106 per well) were inoculated into 6 cm culture plates and incubated in 5% CO2/air at 37 °C for 6 h. Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum (FBS) and 10 mL/L penicillin-streptomycin (5 000 IU/mL and 5 000 mg/mL, respectively; Gibco, USA) was used as the culture medium. The cells were transiently transfected with PPRE, PPARγ, and RXRα expression vectors using FuGene 6. Sixteen hours after transfection, the cells were inoculated into 96-well plates, and following 2 h incubation in 5% CO2/air at 37°C, the test compounds were added and the cells were further incubated for 48 h. Lucifer-ase assay substrate (Promega, USA) was introduced to each well and the intensity of emitted luminescence was determined using EnVision (PerkinElmer, USA). PPARγ trans- criptional activity was expressed as the relative luminescence intensity to that of the control. For antagonist effects, the test compounds were added to the cells 30 min before the addition of BRL49653 (1 µmol/L) and incubated for 24 h. Luciferase activity was determined as above.
Adipogenesis assay 3T3-L1 cells (20 000 cells/well) were planted onto 24-well culture plates and maintained for 2 d after reaching confluence. The culture medium (DMEM, Gibco, USA) was then changed with the differentiation medium (DMEM containing 10% FBS, 1 µmol/L dexamethasone, and 1 µg/mL insulin). The cells were treated with BRL49653 (0.5 µmol/L) or the test compounds (5 µmol/L) for 2 d. Following the change of the medium containing the compounds, the cells were cultured for an additional 3 d. The differentiation medium was replaced by the adipocyte growth medium (DMEM supplemented with 10% FBS and 1 µg/mL insulin) and incubated for a further 2 d. The cells were washed with phosphate-buffered saline and lysed by 0.1% Nonidet P-40 (Sigma-Aldrich, USA). The supernatant of the preparation was examined by the TG detection kit at a wavelength of 500 nm on VERSAmax (Molecular Devices, Sunnyvale, CA, USA).
Data analysis Data were analyzed using GraphPad Prism software (GraphPad, San Diego, CA, USA). Non-linear regression analyses were performed to generate dose–response curves. Ki values were calculated from the half maximal inhibitory concentration (IC50) using the equation of Cheng and Prusoff[15]:
Results
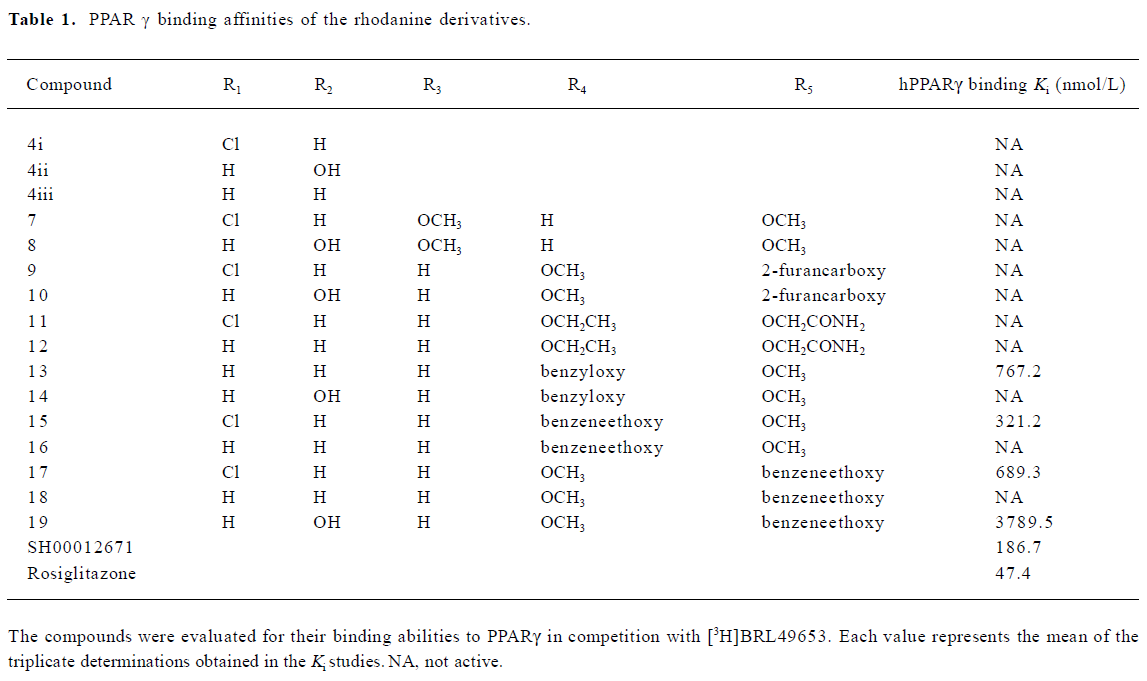
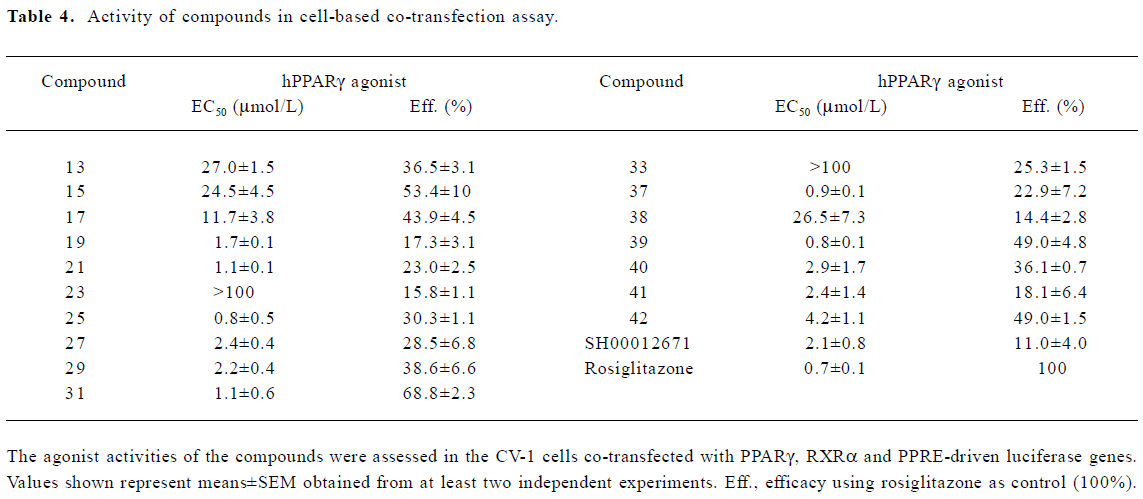
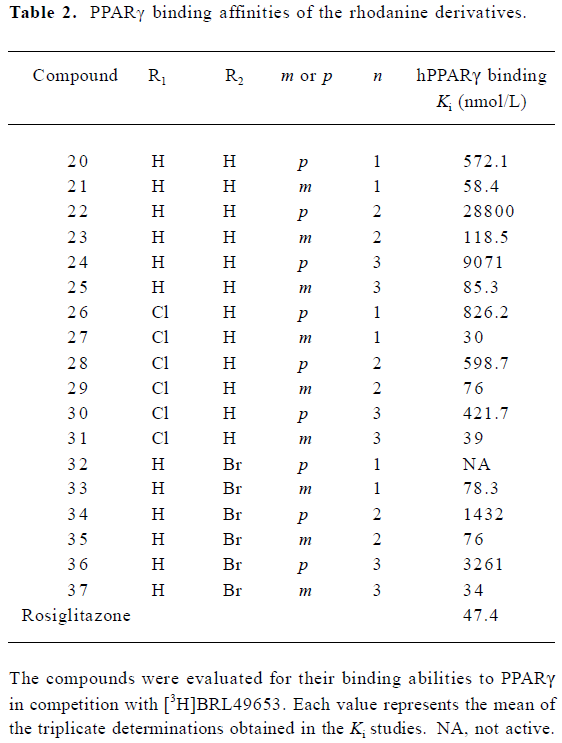
A group of 16 analogues to SH00012671 were synthesized initially (Figure 1) with various substitutions at R1–R5, of which only compounds 13, 15, and 17 showed submicromolar binding affinities to PPARγ, but not as potent as the initial hit (Table 1). However, they displayed some agonist activities with less than 50% efficacies when compared to rosiglitazone in the cotransfection assay (see Table 4 below). We next synthesized another group of 18 rhodanine derivatives by making small modifications on the R1 or R2 (substitution of H with Cl or Br in meta- or para-site) and with different chain lengths. In this case (Table 2), significant improvement in PPARγ binding was observed among one-half of these compounds (21, 23, 25, 27, 29, 31, 33, 35, and 37), to the extent that some of them displayed Ki values similar to that of rosiglitazone (21, 27, 31, and 37). In the cotransfection assay, compounds 21, 25, 31, and 37 exhibited similar half maximal effective concentration (EC50) values compared to rosiglitazone with lower efficacies (23%–69%; see Table 4 below). When tested in the antagonist mode, compounds 27, 31, 29, and 37, with good binding affinities and modest transactivation efficacies, were unable to suppress luciferase activity induced by rosiglitazone (1 µmol/L).
Full table
Full table
Full table
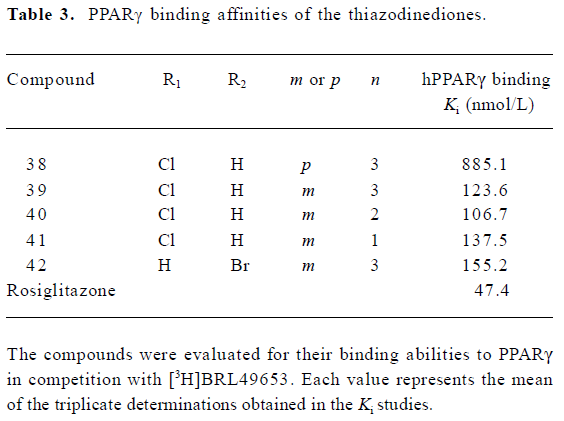
To reduce intrinsic cytotoxicities associated with this class of molecules, the atom thio on the rhodanine was changed to oxygen (Figure 1). This alteration led to a decrease in receptor-binding affinities (Table 3) while the modified analogues maintained efficacies in the cotransfection assay, with the exception of compound 42 (42 vs 37; Table 4).
Full table
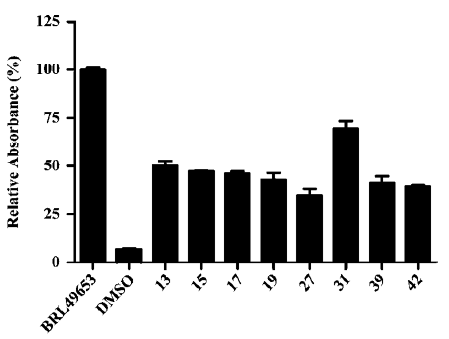
Representative compounds from each of the above 3 groups were evaluated in the pre-adipocyte differentiation assay. All of the 8 compounds tested were able to induce adipogenesis in 3T3-L1 cells with compound 31 being the best inducer relative to BRL49653, consistent with its performance in both the PPARγ-binding and cotransfection assays (Figure 2; Tables 2, 4).
Discussion
In a previous study[12], we reported the discovery of 12 hit compounds with selective PPARγ binding properties, of which 2 belong to the thiazodinedione class, including SH00012671 with a relatively lower IC50 value (0.2 µmol/L). When tested for its effects on cells, it acted either as a poor activator in the cotransfection assay (Table 4) or was inactive in stimulating pre-adipocyte (3T3-L1) differentiation. We thus started some limited chemistry efforts by modifying the rhodanine core structure to better understand the molecular basis of this discrepancy. Three compounds synthesized initially (4i, 4ii, and 4iii) did not bind to PPARγ at all (Table 1), suggesting that benzylidene is required for receptor-binding properties. Therefore, the 5-substituted benzylidene group was introduced, and simple substitutions at R3, R4 and R5 with methoxy, ethoxy, 2-furancarboxy, or aminocarbonyl-methoxy did not change the binding situation (Table 1). How-ever, when R4 or R5 was substituted with benzyloxy or benzeneethoxy, 4 derivatives (13, 15, 17, and 19) possessed both receptor-binding and PPARγ agonist properties (Tables 1, 4). Hydroxy in R2 (14) abolished the receptor-binding ability of 13, while chloride in R1 may be responsible for PPARγ agonist activity (15 vs 16 and 17 vs 18; Tables 1, 4). Cytotoxicity was noted in the cells treated with 19 (data not shown) and this might have led to the lower agonist efficacy observed (Table 4).
In the receptor-binding assay, we found that the removal of methoxy in 5-substituted benzylidene would markedly improve binding activity (Table 2). Among the 18 compounds in this group, meta-substitution behaved better than para-substitution (21 vs 20, 23 vs 22, 25 vs 24, 27 vs 26, 29 vs 28, 31 vs 30, 33 vs 32, 35 vs 34, and 37 vs 36; Table 2). The chain length also affected the receptor-binding activity. Compounds with n=1 or 3 in the chain length bound to PPARγ more potently than compounds with n=2 in the chain length (21, 25 vs 23 and 27, and 31 vs 29; Table 2). When R1 or R2 was substituted with halogen, the receptor-binding activity was also improved (27 vs 21; 29, 35 vs 23; 31, and 37 vs 25; Table 2). In the PPARγ cotransfection assay, both the chain length and halogen substitution affected the agonist properties. The activities of compounds with n=3 in the chain length were better than compounds with n=1 or 2 in the chain length (25, 31, 37, and 39; Table 4). The halogen (eg Cl) group also increased the cotransfection activity (27, 29, and 31; Table 4).
To reduce intrinsic cytotoxicities, the atom thio on the rhodanine was changed to oxygen. This alteration led to a decrease in receptor-binding affinities while modified analogues generally maintained efficacies in cell-based assays, except compound 42 (42 vs 37; Table 4). The underlining mechanism of this phenomenon deserves further investi-gation.
According to Amit and colleagues[16], a 3-D quantitative structure–activity relationship model of PPARγ agonist thiazolidine-2,4-dione has 3 biophoric centers, A, B, and C, corresponding to carbonyl oxygen of thiazolidine-2,4-dione, sulphur atom of thiazolidine-2,4-dione, and the oxygen atom attached to the phenyl ring, respectively. These are electron-rich sites capable of donating electrons and may be involved in electrostatic, ionic, and π-π interactions. Site C is essential to the agonistic action of PPARγ, whereas the carbonyl group of thiazolidinediones (site A) may be responsible for the formation of hydrogen bonds with 2 histidine residues, His 323 and 449, of PPARγ. This information could certainly help us understand why the benzyloxy, phenyl-ethoxy, and phenylpropoxy groups are important to the PPARγ agonists described in the present study.
It is conceivable that the modest transactivation efficacies observed with these compounds (68.8% maximum compared to rosiglitazone) were due to partial agonist effects. The fact that they did not behave as antagonists in the same assay system suggests that their potencies at PPARγ should be further improved. Likewise, we noted some inconsistencies relative to transactivation versus adipogenesis assays in terms of agonist effects. Although some agonists have relatively potent agonistic activities in the cotransfection assay (eg 39), they acted poorly on pre-adipocyte differentiation. This is in agreement with previous studies on a group of PPARγ agonists, including KR-62980, GW0072, FMOC-L-leucine, PAT5A and nTZDpa[17]. Biochemical and structural studies with PPARγ revealed that different ligands may occupy the PPARγ ligand-binding pocket in different manners, thereby inducing different allosteric changes in its conforma-tion. Such alterations would result in different kinds of interactions between receptor and cofactor proteins, such as steroid receptor co-activator 1 (SRC1), nuclear receptor co-activator TIF2, cAMP response element (CRE) binding protein (CBP), TRAP220/DRIP205, nuclear receptor co-repressor (NcoR), and the silencing mediator of the retinoid and thyroid hormone receptors (SMRT)[18]. Therefore, the effect of PPARγ modulators on the downstream adipogenesis and glucose uptake will depend on the context of interaction between the ligand and the receptor.
In summary, 5-benzylidene was required for the receptor-binding properties. Simple substitutions on benzylidene with methoxy, ethoxy, 2-furancarboxy, or amino-carbonyl-methoxy did not change the binding affinity. However, when benzylidene was substituted with aralkyl, both receptor-binding and PPARγ agonist activities were demonstrable. Meta-substitution was better than that of para. The chain length also affected the PPARγ-binding and agonist activities. The best chain length studied was 3. Finally, the substitution of halogen in R1 or R2 improved binding activity. Of the analogues studied, compound 31 exhibited about 70% the efficacy exerted by BRL49653 in both the cotransfection and pre-adipocyte differentiation assays, in addition to its weak agonist action on hPPARα (data not shown). High receptor binding potency (Ki =47.4 nmol/L) and less than optimal agonist efficacy (EC50=700 nmol/L) relative to rosiglitazone create an opportunity to use rhodanine derivatives as a new scaffold in further understanding the molecular mechanism of agonism at PPARs.
Acknowledgements
We are indebted to Xin HUI and Meng-meng NING for technical assistance.
References
- Wilson TM, Wahli W. Peroxisome proliferators-activated receptor agonists. Curr Opin Chem Biol 1997;1:235-41.
- Braissant O, Foufelle F, Scotto C, Dauca M, Wahli W. Differential expression of peroxisome proliferator-activated receptors (PPARs): tissue distribution of PPAR-α, -β, and -γ in the adult rat. Endocrinology 1996;137:354-66.
- Kliewer SA, Forman BM, Blumberg B, Ong ES, Borgmeyer U, Mangelsdorf DJ, et al. Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc Natl Acad Sci USA 1994;91:7355-9.
- Kliewer SA, Umesono K, Noonan DJ, Heyman RA, Evans RM. Convergence of 9-cis retinoic acid and peroxisome proliferator signaling pathways through heterodimer formation of their receptors. Nature 1992;358:771-4.
- Ijpenberg A, Jeannin E, Wahli W, Desvergne B. Polarity and specific sequence requirements of PPAR-RXR heterodimer binding to DNA: a functional analysis of the malic enzyme gene PPRE. J Biol Chem 1997;272:20108-17.
- Aubry CJ, Pernin A, Favez T, Burger AG, Wahli W, Meier CA, . DNA binding properties of peroxisome proliferator-activated receptor subtypes on various natural peroxisome proli-ferator response elements: importance of the 5’ flanking region. J Biol Chem 1997; 272: 25 252–9.
- Keller JK, Collet P, Bianchi A, Huin C, Kremarik PB, Becuwe P, et al. Implications of peroxisome proliferator activated receptors (PPAR) in development, cell life status and disease. Int J Dev Biol 2000;44:429-42.
- Sohda T, Momose Y, Meguro K, Kawamatsu Y, Sugiyama Y, Ikeada H. Studies on antidiabetic agents. Synthesis and hypoglycemic activity of 5-[4-(pyridylalkoxy)benzyl]-2,4-thiazolidine-diones. Arzneimittelforschung 1990;40:37-42.
- Kletzien RF, Clarke SD, Ulrich RG. Enhancement of adipocyte differentiation by an insulin-sensitizing agent. Mol Pharmacol 1992;41:393-8.
- Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferators-activated receptor gamma (PPARγ). J Biol Chem 1995; 270: 12 953–6.
- Javiya VA, Patel JA. The role peroxisome proliferator-activated receptors in human disease. Edu Forum 2006; 38: 243–53.
- Wu B, Gao J, Wang MW. Development of a complex scintillation proximity assay for high-throughput screening of PPARγ modulators. Acta Pharmacol Sin 2005;26:339-44.
- Gearing KL, Göttlicher M, Teboul M, Widmark E, Gustafsson J. Inter-action of the peroxisome proliferator-activated receptor and retinoid X receptor. Proc Natl Acad Sci USA 1993;90:1440-4.
- Tadashi S, Masaki O. Studies on sulfur-containing heterocyclic compounds. II. Reaction of potassium 3-benzoyldithiocarbazate and monochloroacetic acid and its ester. J Pharm Soc Japan 1955;75:1535-9.
- Cheng Y, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (IC50) of an enzymatic reaction. Biochem Pharmacol 1973;22:3099-108.
- Amit K, Sushil KK, Anil KS. QSAR and molecular modeling studies in imidazopyridinethiazolidine-2,4-diones: PPARγ agonists. Med Chem Res 2004;13:770-80.
- Kwang RK, Jeong HL, Seung JK, Sang DR, Won HJ, Sung-Don Y, et al. KR-62980: A novel peroxisome proliferator-activated receptor γ agonist with weak adipogenic effects. Biochem Pharmacol 2006;72:446-54.
- Béatrice D, Walter W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev 1999;20:649-88.