
Novel selective inhibitors of hydroxyxanthone derivatives for human cyclooxygenase-2
Introduction
The prostaglandin endoperoxide H synthase (PGHS), cyclooxygenase (COX), is in charge of prostaglandin synthesis[1]. Arachidonic acid (AA) is catalyzed into prostaglandin H2 (PGH2) through the dual functions of PGHS[2]. Several prostanoids, which maintain normal physiologies, are metabolized from the precursor PGH2[3,4]. Two isozymes have been identified: COX-1 is generally expressed in most tissues and cells, and the production of COX-1 is maintain several physiological functions[5,6]; COX-2 is inducible by growth factors, cytokines, and endotoxins[7], and the prostanoids act as the potent mediators of fever and inflammation[8]. At present, many clinical reports have found that tumorigenesis of some certain tissues and cells is correlated with COX-2[9–11]. COX-2 has been found to induce angiogenic factors regulated angiogenesis[10,11], specifically in colorectal cancer cells. Some carcinogenesis could be reduced by inhibiting the overexpression of COX-2. For centuries, non-steroidal anti-inflammatory drugs (NSAIDs)[12] have been taken as inhibitors of COX-2 to ease pain[13]. These drugs even showed the efficiency to decrease incidence of esophageal, gastric, and colorectal cancers[14,15]. The two isozymes, COX-1 and COX-2 have high structure similarity (root mean square difference, RMSD <1.0 Å) and approximately 60% identity for shared residues[16]. Furthermore, with an emphasis on the similarity of active sites, there are only 2 different amino acids between the isoforms, His513 and Ile523 of human COX-1, and Arg499 and Val509 of human COX-2[17]. According to the above reasons, most NSAIDs are not selective for both isozymes. A consequence is several side-effects, which is as a result of the biosynthesis being inhibited[18]. Several studies have reported that long-term aspirin usage might induce stomach ulcers[19–21]. A strategy of developing novel drugs with higher selectivity is important, and the efficacy and side-effects should be considered.
The commercial compounds, resveratrol and NS-398, are known specific inhibitors of COX-1 and COX-2, respectively[22,23]. Resveratrol and NS-398 were recruited as the control compounds for this study. The xanthone derivatives were extracted from Garcinia mangostana (mangosteen) and have several biological activities, including the effect of anti-inflammatory, anti-bacterial, and skin-infection free[24]. These derivatives were used reasonably against the inflammatory function of COX-2. Furthermore, one of the derivatives shows a potent cytotoxic effect against hepatoma[25]. The derivatives, α-mangostin and γ-mangostin, have the ability of preventing AA from binding to the active sites of both isozymes[26–28]. In this study, hydroxyxanthones were investigated. A further structure-based research methodology of virtual screening was employed to analyze the selectivity of the 2 isozymes. The program LigandFit (Accelrys, San Diego, CA, USA) was used for the docking procedures[29]. Several scoring functions, including dock score, piecewise linear potential (PLP)[30], and potential of mean force (PMF)[31], were taken to evaluate the docking results. The dock score is a minus value of the summation of ligand/receptor interaction energy and ligand internal energy. By considering 4 PLP atom-type predefinitions, the PLP score indicates the sums of all atomic pairwise interactions of the ligand/receptor complex. The PMF calculates all the interatomic pairs of the ligand/receptor complex. Because the values of PLP and PMF are minus, it would be added a minus prefix before each the indicator, ie -PLP and -PMF. A higher score may suggest a higher affinity, as the values of energy would be presented with the sign reversed. The interaction energy between ligand and receptor was calculated under the force field at the Department of Chemistry at Harvard Macromolecular Mechanics (CHARMm) according to the following equation[32]:
Einteraction=Ecomplex–(Eligand+Ereceptor),
E: energy.
These calculations of scoring functions and interaction energy represented the purpose of evaluating the ligand and protein interactions. It might suggest the prediction of selectivity to both isozymes and the directions of further rational drug design.
Materials and methods
Homology modeling Molecular simulation was performed under Discovery Studio modeling 1.7 (Accelrys, San Diego, CA, USA). The protein templates were obtained from Protein Data Bank (PDB). Ovine prostaglandin H2 synthase-1 in complex with alpha-methyl-4-biphenylacetic acid (Ovis aries; PDB ID: 1Q4G)[33], and AA bound to the COX-active site of COX-2 (Mus musculus; PDB ID: 1CVU)[34]. The sequences of human COX-1 (Swiss Prot accession N
Docking prediction and interaction energy calculation The simulated structure was defined as “SBD_RECEPTOR” in the program, and the cavities as the binding sites within the receptor were discovered by computational prediction corresponding to the known active site within the key residues, Arg120, Leu352, Tyr355, Tyr348, Trp387, Ser530, and Leu 531 for COX-1[35]. For the same residues of human species, actually the residue numbers should decrease one residue number based on ovine species and distributed over the same space in each structure, such as Arg120 to Arg119. Xanthone derivatives were prepared by Chem Office 2005 software (Cambridge Scientic Computmg, Cambridge, Massachusetts, USA), including sketch and energy minimization (MM2 force field)[36]. These derivatives, compounds A–H, were employed as the ligands while performing the docking process. NS-398 and resveratrol were recruited as the controls. By using LigandFit, compounds A–H were docked into a cavity, which was located in the active site. All the ligands were flexible in the docking procedure, while the receptor was fixative. The poses of all the compounds were arranged by the LigandFit module in the active site by Monte Carlo trials, and the dock score was calculated relying on the generated pose by the Dreiding energy grid force field. Scoring functions, PLP, and PMF, scored the docking results for each compound within the receptor. After docking, the energies of the ligand/receptor complexes were minimized. Then the interaction energy of each complex was calculated according to the premised equation.
Results
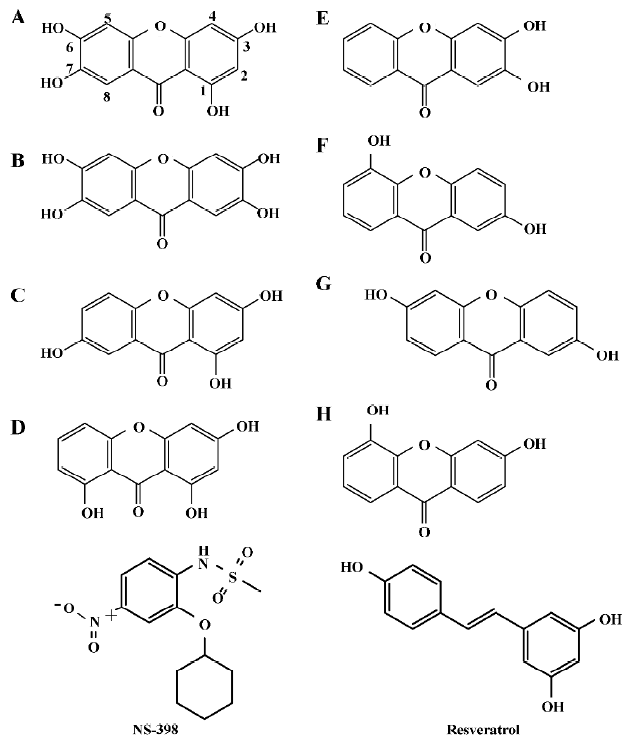
Structure modeling and analysis of xanthone derivatives Both human isozymes were built under comparative modeling. The sequence identity between the template and the target protein for COX-1 and COX-2 was 93.9% and 87.6%, respec-tively. With the high similarity and identity of the protein templates, the built protein structures were well folded. According to the Ramachandron plot and Verify3D of each crystal and simulated structures, the amino acids of the outer regions were located at the peripheral residues and anchor motif (data not shown). The anchor motif was far from the active site, so it was reasonable to suppose that there was no influence on the mechanism of AA catalyzing. All the derivatives employed in this study are represented in Figure 1. There were 2 tetrahydroxyxanthones, 2 trihydroxyxan-thones, and 4 dihydroxyxanthones. The compound A, 1,3,6,7-tetrahydroxyxanthone, revealed some similarities with garcinone E. Compared with another tetrahydroxyxanthone, compounds A and B indicate a wider structure with the meta-hydroxyl group than the ortho-hydroxyl group. Compound C present as a narrower structure than compound A. Compound D is shorter in the length than compound C. Half of the hydroxyl groups are on compound E than on compound B.
Binding-site definition and scoring function evaluation The predictive binding sites of COX-1 and COX-2 were matched with the actual active sites, respectively. A further interaction analysis was conducted following the study of the structure-based design. The scoring functions showed the ranking of each compound in the fixed receptor. By considering the scores and poses of all the compounds in the lobby of the protein, better ligands were obtained. The interactivon energy between ligand and receptor was considered after compound screening, while lower interaction energy indicated a more stable binding status.
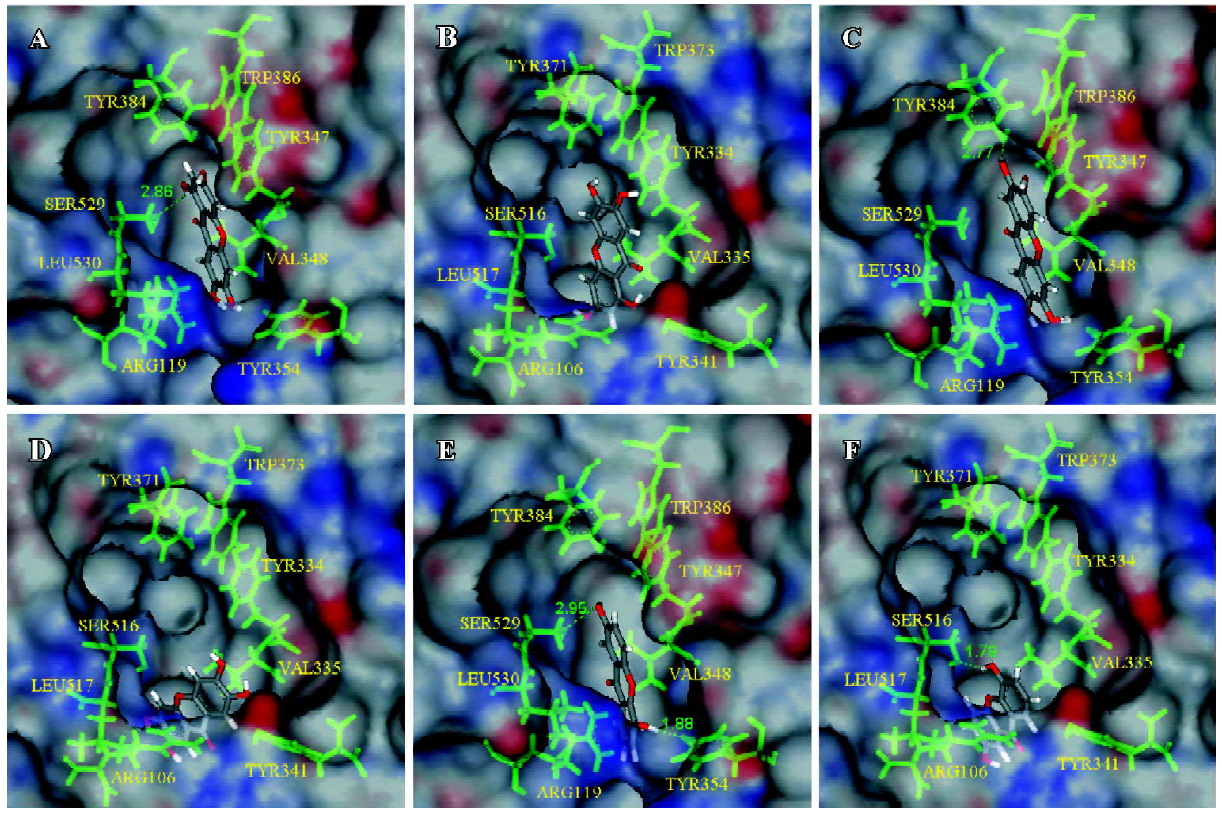
Docking results and interaction energy calculations of conformations of the marked ligands Compound A showed a higher interaction force with both COX isozymes (Table 1). The scoring functions in Table 1 suggest that the scaffold of 1,3,6,7-hydroxyxanthone interacted well with the active site. Notably, compound A bound to Ser529, as COX-1 was similar to aspirin which could acetylate Ser529 to prevent the binding of AA[32,37]. For lower interaction energy, compound A might be more stable than resveratrol and NS-398 in the active sites of COX-1 and COX-2, respectively. The docking pose of NS-398 was similar to SC-558 in another protein template (PDB ID: 6COX). In addition, the simulation results of these 2 control compounds, resveratrol and NS-398, corresponded well with biological data. We suggest that the simulation protein structures (human COX-1 and COX-2) and all the simulation docking results were reliable. The docking results of compounds A, B, and F, and the receptors were displayed through molecular simulation as shown in Figure 2. It could be deduced that compound A might have better inhibitory ability to both isoforms. From our observations, we found that compound B had more inhibitory potency to COX-1 than COX-2 through the calculation of interaction energy. The active site of COX-1 showed a tunnel-like structure, while the site of COX-2 was roomier than COX-1. With the scoring functions showing the same tendency, it was indicated that the agents with slimmer structures were more suitable in COX-1 than in COX-2. Compound B revealed an interesting result; the docking poses were different between the complexes of compound B in COX-1 and COX-2. The docking pose of compound B was like the pose of the docked compound A and it gained a higher dock score and a more stable energy status (Figure 2C). Although the docking conformation of compound B had no hydrogen bond predicted, it might be steadier in the space around the residues than the pose in Figure 2D. Compounds C and F showed potent selectivity to COX-2 with obvious differences in the interaction energy (188.57 and 215.56 kJ/mol), respectively. Through the structure-based study, compound F was shown to have a hydrogen bond with Ser529 in COX-1, as indicated in Figure 2E, while the horizontal orientation of compound F in the active site of COX-2 showed that the pose in Figure 2F resembled the conformation of compound B in both COX-1 and COX-2. One strong hydrogen bond with 1.79 Å was observed in the compound F/COX-2 complex. Depending on this conformation, compound F could achieve lower interaction energy and the higher dock score within the receptor. It could be suggested that compound F might be a potent novel lead compound for COX-2. Additionally, compounds D, E, G, and H obtained stable statuses while they docked with COX-2 than with COX-1. The conformations of D, E, and G were similar, with compound F in the active site of both protein structures (Figure 2E, 2F), respectively.
Full table
Discussion
By considering the scoring functions, a better conformation of compounds could be ranked. These conformations influenced the integral stability of the ligand and receptor complex, but the stability of energy within the ligand/receptor complex would be the major emphasis of real time. Lower interaction energy would imply more stable ligand/receptor complexes.
According to structure-based researching methodology, the feasible prediction of binding sites was calculated, and every molecular docking result was analyzed through the scoring functions, interaction energy, and structure con-formation. Compounds A, C, and D showed lower interaction energy compared to NS-398 in COX-2. Compound B revealed selectivity with COX-1 and lower interaction energy than resveratrol in COX-1. Notably, compound A, D, and H also had better binding forces than resveratrol. It is interesting to investigate the docking poses of compounds B and E because of the difference in 2 of the hydroxyl groups. Compound B could bind to the gate of active site, Arg119 and Tyr354, of COX-1 counting on the two more hydroxyl groups and thus gaining more stable than compound E in the lobby. Furthermore, the different hydroxyl groups between compounds F and G changed the results slightly. The binding poses were added in the supplementary part. It was noticed that compound G in the active site of COX-1 gained 1 hydrogen bond with Tyr354 by its 5'-hydroxyl group. For the adaption of this pose, the 2'-hydroxyl group of compound G bound to Tyr384 instead of Ser529 because the scaffold of this ligand docked with the upper position than compound F in COX-1.
Compound A bound well to both isozymes by LigandFit. Compound B showed selective potency of COX-1, while compound F showed selectivity to COX-2 through structure-based research. Because the cavity of the COX-2 catalytic site was larger than COX-1, these non-shape-specific compounds were docked into the site non-specifically. Due to this reason, the interaction energy varied with different docking poses. Further pharmacophores should be generated for the purpose of studying a quantitative structure–activity relationship between the complexes, and the design of more efficient inhibitors should be the next step.
References
- Li L, Pettit AR, Gregory LS, Forwood MR. Regulation of bone biology by prostaglandin endoperoxide H synthases (PGHS): a rose by any other name. Cytokine Growth Factor Rev 2006;17:203-16.
- Tsai A, Kulmacz RJ, Palmer G. Spectroscopic evidence for reaction of prostaglandin H synthase-1 tyrosyl radical with arachidonic acid. J Biol Chem 1995; 270: 10 503–8.
- Kulmacz RJ. Regulation of cyclooxygenase catalysis by hydroperoxides. Biochem Biophys Res Commun 2005;338:25-33.
- Shimamoto C, Nakanishi Y, Katsu K, Nakano T, Kubota T, Mori H, et al. Prostaglandin E2 release in gastric antral mucosa of guinea-pigs: basal PGE2 release by cyclo-oxygenase 2 and ACh-stimulated PGE2 release by cyclo-oxygenase 1. Exp Physiol 2006;91:1015-24.
- Konturek PC, Konturek SJ, Bielanski W, Kania J, Zuchowicz M, Hartwich A, et al. Influence of COX-2 inhibition by rofecoxib on serum and tumor progastrin and gastrin levels and expression of PPARgamma and apoptosis-related proteins in gastric cancer patients. Dig Dis Sci 2003;48:2005-17.
- Zhang WY, Yang XN, Jin DZ, Zhu XZ. Expression and enzyme activity determination of human cyclooxygenase-1 and -2 in a baculovirus-insect cell system. Acta Pharmacol Sin 2004;25:1000-6.
- Stoll G, Bendszus M. Inflammation and atherosclerosis: novel insights into plaque formation and destabilization. Stroke 2006;37:1923-32.
- Chen XH, Bai JY, Shen F, Bai AP, Guo ZR, Cheng GF. Imrecoxib: a novel and selective cyclooxygenase 2 inhibitor with anti-inflammatory effect. Acta Pharmacol Sin 2004;25:927-31.
- Kundu N, Fulton AM. Selective cyclooxygenase (COX)-1 or COX-2 inhibitors control metastatic disease in a murine model of breast cancer. Cancer Res 2002;62:2343-6.
- Chen PY, Long QC. Effects of cyclooxygenase 2 inhibitors on biological traits of nasopharyngeal carcinoma cells. Acta Pharmacol Sin 2004;25:943-9.
- Tsujii M, Kawano S, Tsuji S, Sawaoka H, Hori M, DuBois RN. Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell 1998;93:705-16.
- Lora M, Denault JB, Leduc R, de Brum-Fernandes AJ. Systematic pharmacological approach to the characterization of NSAIDs. Prostaglandins Leukot Essent Fatty Acids 1998;59:55-62.
- Stone E. An account of the success of the bark of the willow in the cure of agues. Phil Trans 1763;53:195-200.
- Hao LL, Mei QB, Zhang BL, Jia M, Li XQ, Zhang F. PC-407 inhibited proliferation and induced apoptosis in human colon cancer SW-1116 cells. Acta Pharmacol Sin 2004;25:1509-14.
- Reddy BS, Hirose Y, Lubet R, Steele V, Kelloff G, Paulson S, et al. Chemoprevention of colon cancer by specific cyclooxygenase-2 inhibitor, celecoxib, administered during different stages of carcinogenesis. Cancer Res 2000;60:293-7.
- Kurumbail RG, Kiefer JR, Marnett LJ. Cyclooxygenase enzymes: catalysis and inhibition. Curr Opin Struct Biol 2001;11:752-60.
- Wong E, Bayly C, Waterman HL, Riendeau D, Mancini JA. Conversion of prostaglandin G/H synthase-1 into an enzyme sensitive to PGHS-2-selective inhibitors by a double His513 right-arrow Arg and Ile523 right-arrow val mutation. J Biol Chem 1997;272:9280-6.
- Li XH, Li JJ, Zhang HW, Sun P, Zhang YL, Cai SH, et al. Nimesulide inhibits tumor growth in mice implanted hepatoma: overexpression of Bax over Bcl-2. Acta Pharmacol Sin 2003;24:1045-50.
- Colin-Jones DG. Drug-induced gastrointestinal ulceration: a review. J R Soc Med 1980;73:46-7.
- Laine L, Maller ES, Yu C, Quan H, Simon T. Ulcer formation with low-dose enteric-coated aspirin and the effect of COX-2 selective inhibition: a double-blind trial. Gastroenterology 2004;127:395-402.
- Niederberger E, Manderscheid C, Geisslinger G. Different COX-independent effects of the COX-2 inhibitors etoricoxib and lumiracoxib. Biochem Biophys Res Commun 2006;342:940-8.
- Yoshimi N, Shimizu M, Matsunaga K, Yamada Y, Fujii K, Hara A, et al. Chemopreventive effect of N-(2-cyclohexyloxy-4-nitro-phenyl)methane sulfonamide (NS-398), a selective cyclooxygen-ase-2 inhibitor, in rat colon carcinogenesis induced by azoxy-methane. Jpn J Cancer Res 1999;90:406-12.
- Szewczuk LM, Forti L, Stivala LA, Penning TM. Resveratrol is a peroxidase-mediated inactivator of COX-1 but not COX-2: a mechanistic approach to the design of COX-1 selective agents. J Biol Chem 2004; 279: 22 727–37.
- Matsumoto K, Akao Y, Kobayashi E, Ohguchi K, Ito T, Tanaka T, et al. Induction of apoptosis by xanthones from mangosteen in human leukemia cell lines. J Nat Prod 2003;66:1124-7.
- Ho CK, Huang YL, Chen CC. Garcinone E, a xanthone derivative, has potent cytotoxic effect against hepatocellular carcinoma cell lines. Planta Med 2002;68:975-9.
- Moongkarndi P, Kosem N, Kaslungka S, Luanratana O, Pongpan N, Neungton N. Antiproliferation, antioxidation and induction of apoptosis by Garcinia mangostana (mangosteen) on SKBR3 human breast cancer cell line. J Ethnopharmacol 2004;90:161-6.
- Nabandith V, Suzui M, Morioka T, Kaneshiro T, Kinjo T, Matsumoto K, et al. Inhibitory effects of crude alpha-mangostin, a xanthone derivative, on two different categories of colon preneoplastic lesions induced by 1, 2-dimethylhydrazine in the rat. Asian Pac J Cancer Prev 2004;5:433-8.
- Nakatani K, Nakahata N, Arakawa T, Yasuda H, Ohizumi Y. Inhibition of cyclooxygenase and prostaglandin E2 synthesis by gamma-mangostin, a xanthone derivative in mangosteen, in C6 rat glioma cells. Biochem Pharmacol 2002;63:73-9.
- Venkatachalam CM, Jiang X, Oldfield T, Waldman M. LigandFit: a novel method for the shape-directed rapid docking of ligands to protein active sites. J Mol Graph Model 2003;21:289-307.
- Gehlhaar DK, Verkhivker GM, Rejto PA, Sherman CJ, Fogel DB, Fogel LJ, et al. Molecular recognition of the inhibitor AG-1343 by HIV-1 protease: conformationally flexible docking by evolutionary programming. Chem Biol 1995;2:317-24.
- Muegge I, Martin YC. A general and fast scoring function for protein-ligand interactions: a simplified potential approach. J Med Chem 1999;42:791-804.
- Selvam C, Jachak SM, Gnana Oli R, Thilagavathi R, Chakraborti AK, Bhutani KK. A new cyclooxygenase (COX) inhibitory pterocarpan from Indigofera aspalathoides: structure elucidation and determination of binding orientations in the active sites of the enzyme by molecular docking. Tetrahedron Lett 2004;45:4311-4.
- Gupta K, Selinsky BS, Kaub CJ, Katz AK, Loll PJ. The 2.0 A resolution crystal structure of prostaglandin H2 synthase-1: structural insights into an unusual peroxidase. J Mol Biol 2004;335:503-18.
- Kiefer JR, Pawlitz JL, Moreland KT, Stegeman RA, Hood WF, Gierse JK, et al. Structural insights into the stereochemistry of the cyclooxygenase reaction. Nature 2000;405:97-101.
- Filizola M, Perez JJ, Palomer A, Mauleon D. Comparative molecular modeling study of the three-dimensional structures of prostaglandin endoperoxide H2 synthase 1 and 2 (COX-1 and COX-2). J Mol Graph Model 1997;15:290-300.
- Allinger NL. Conformational analysis 130.MM2. A hydrocarbon force field utilizing V1 and V2 torsional terms. J Am Chem Soc 1977;99:8127-34.
- De Witt DL. Cox-2-selective inhibitors: the new super aspirins. Mol Pharmacol 1999;55:625-31.