
AICAR inhibits proliferation and induced S-phase arrest, and promotes apoptosis in CaSki cells1
Introduction
Cervical cancer is the second leading cause of cancer morbidity and mortality for women around the world, especially in many developing countries[1]. The aberrancy of signaling pathways is critical in the pathogenesis and progression of cancers. Thus far, many signaling transduction components have been explored as potential therapeutical targets in cervical cancer, such as p53[2], extracellular signal-regulated kinase (ERK)[3], phosphatidylinositol 3-kinase (PI3K)[4], and mammalian target of rapamycin (mTOR)[5].
5-Aminoimidazole-4-carboxamide-ribonucleoside (AICAR) is a well-known activator of the adenosine mono-phosphate(AMP)-activated protein kinase (AMPK), which has been proposed to act as a cellular fuel sensor[6]. AMPK is activated when the cellular AMP/ATP ratio increases. Activated AMPK inactivates the ATP-consuming cellular events and activates ATP-generating processes. Recently, AMPK was reported to be involved in cell growth regulation and was identified as a potential target for cancer[7]. It has a close relationship with 2 tumor suppressor genes: LKB1 and tuberous sclerosis complex 2[6], which are important negative regulators of cell size and growth. It was reported that AICAR could inhibit the proliferation of several cancer cell lines, such as C6 glioma cells, T98 G astrocytoma cells, MCF-7 breast cancer cells, PC-3 prostate carcinoma cells, and hematological cancer cells[8,9]. AICAR could also upregulate the expression of tumor suppressor p53 in some of those cancer cell lines[8] and can inhibit mTOR signaling[10]. Recently, Nafz et al reported that AICAR could also induce human papillomaviruses (HPV) suppression in cervical cancer cell lines[13].
The aim of the present study is to elucidate the role of AICAR in cervical cancer cells. We treated cervical cancer cell line CaSki with AICAR at different concentrations to dynamically observe the effect of AICAR on cell proliferation, cell cycle, and apoptosis. In addition, we also explored the effect of AICAR on some important kinases associated with growth control.
Materials and methods
Reagents Human cervical epithelial cell line CaSki was obtained from the American Type Culture Collection (Manassas, VA, USA). AICAR was obtained from Toronto Research Chemicals (Toronto, Canada). Fetal bovine serum (FBS) was from Hyclone (Logan, UT, USA). The Annexin-V kit was purchased from Biosea Biotechnology (Beijing, China). Phospho-ERK (phospho-Thr202/Tyr204), phospho-AKT (Ser473), AKT (also known as protein kinase B), and phospho-mTOR (Ser2448) were from Cell Signaling Technology (Beverly, MA, USA). The antibodies against proliferating cell nuclear antigen (PCNA), p53, caspase-3, ERK, and eukaryotic initiation factor 5(eIF5) were from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Cell culture The CaSki cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% FBS, 100 U/mL penicillin, and 100 µg/mL streptomycin (Life Technologies, Gaithersburg, MD, USA). The cells were cultured at 37 oC in a humidified atmosphere with 5% CO2.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay Exponentially growing cells were seeded at 1×104 cells/well in 96-well plates and incubated with AICAR at various concentrations for 24, 48, and 72 h, respectively. The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, 5 mg/mL; Sigma, St Louis, MO, USA) was added to each well and incubated for 4 h at 37 oC. The formazan product was dissolved by adding 150 µL DMSO to each well. The MTT absorbance value was detected at 490 nm with a micro Sentence plate reader (Bio-Rad, Hercules, CA, USA).
Cell count assay The cells were plated at a density of 5×104/well in 12-well plates in triplicate. DMEM supplemented with 10% FBS was used as the growth medium. After incubation with AICAR (500 µmol/L) for 24–72 h, the cells were harvested and exposed to 0.4% (w/v) trypan blue dye solution; the number of cells that were not stained with trypan blue was counted as viable cells.
Flow cytometry analysis For the cell cycle analysis, approximately 1×106 cells were incubated with AICAR for 24–72 h. The cells were trypsinized and washed twice with cold phosphate-buffered saline (PBS) and then fixed in 70% ethanol at -4 oC overnight. Ethanol-fixed cells were resuspended in PBS containing 0.1 mg/mL RNase and incubated at 37 oC for 30 min. The pelleted cells were suspended in 0.5 mL of 50 µg/mL propidium iodide (PI) and analyzed with a flow cytometer (FACScan, Becton-Dickinson, San Jose, CA, USA). The cell cycle distribution was determined by ModFit LT cell cycle analysis software (Verity Software House, Topsham, ME, USA). For the detection of the percentage of apoptotic cell by Annexin-V/PI, after incubation with AICAR for 24–48 h, the CaSki cells were harvested and washed with cold PBS twice and then resuspended in 200 µL binding buffer at a concentration of 1×106 cells/mL. The cells were incubated with 10 µL Annexin-V–fluorescein isothiocyanate (FITC) and 5 µL PI in the dark for 15 min. In total, 300 µL binding buffer was then added in each tube prior to being analyzed with the flow cytometer.
Analysis of nuclear morphology The cells were seeded in the 6-well plates and treated with AICAR (500 µmol/L) for 24–48 h. After washing twice with PBS, the cells were collected and fixed in methanol and then stained with 20 mg/mL Hoechst 33258 staining solution. Following washing with ice-cold PBS twice, the stained nuclei were examined and immediately photographed under a fluorescence microscope (Leica DMLB, Wetzlar, Germany). Apoptotic cells were defined on the basis of nuclear morphology changes, such as chromatin condensation and fragmentation.
Western blot analysis The CaSki cells were lysed in lysis buffer [20 mmol/L Tris-HCl (pH 7.4), 100 mmol/L NaCl, 10 mmol/L sodium pyrophosphate, 5 mmol/L EDTA, 50 mmol/L NaF, 1 mmol/L sodium vandate, 0.1% SDS, 10% glycerol, 1% Triton X-100, 1% sodium deoxycholate, 1 µmol/L leupeptin, 0.1 µmol/L aprotinin, and 1 mmol/L phenylmethanesulfonyl fluoride (PMSF)]. The protein concentrations were measured by a BCA (Bicichoninic Acid) protein assay kit (Pierce Biotechology, Rockford, IL, USA). The samples (30 µg) were loaded and separated by SDS-PAGE and then electrophoretically transferred to nitrocellulose membranes (Pall, Port Washington, NY, USA). The membranes were probed with various antibodies according to the supplier’s protocol and visualized with peroxidase and an enhanced chemiluminescence system (Pierce Biotechno-logy, IL, USA).
Statistical analysis Data are expressed as mean±SEM. The statistical significance of the differences was determined by ANOVA followed by Student–Newman–Keuls (S–N–K) test or unpaired two-tailed t-tests. A value of P<0.05 was considered significant.
Results
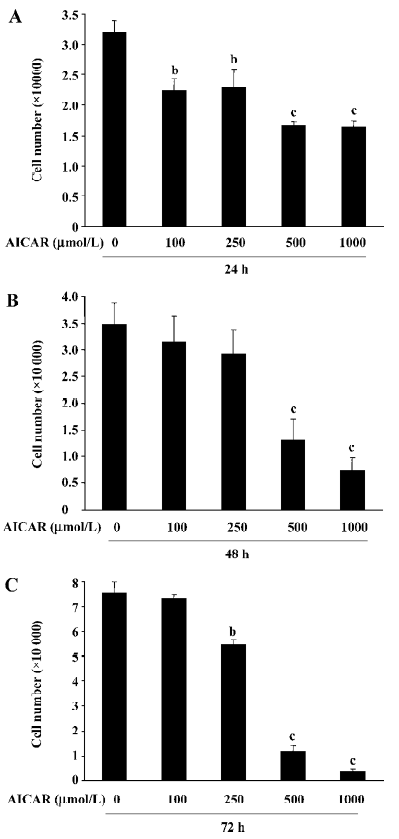
AICAR inhibits the proliferation of CaSki cells To determine the effect of AICAR on the proliferation in CaSki cells, the cells were incubated with AICAR at different concentrations (100, 250, 500, and 1000 µmol/L, respectively) for 24, 48, and 72 h. The number of cells was counted with a hemocytometer. After 24 h of incubation, all doses of AICAR significantly reduced the cell number (Figure 1A). The number of cells decreased from (3.21±0.18)×104 to (2.23±0.19)×104, (2.29±0.31)×104, (1.67±0.06)×104, and (1.64±0.09)×104, respectively. However, 100 µmol/L AICAR did not significantly reduce the cell number when treated for 48 and 72 h. At 48 h, both 500 µmol/L and 1000 µmol/L AICAR significantly decreased the cell number compared with the control [from (3.48±0.40)×104 to (1.31±0.39)×104 and (0.73±0.24)×104] except for 250 µmol/L AICAR which had no statistical effect on the reduction of the cell number (Figure 1B). After incubation for 72 h with AICAR, there was a dose-dependent decrease of the cell number for AICAR at 250, 500, and 1000 µmol/L [from (7.55±0.43)×104 to (5.47±0.15)×104, (1.19±0.21)×104, and (0.41±0.08)×104; Figure 1C]. These results indicated that AICAR inhibited the proliferation of CaSki cells.
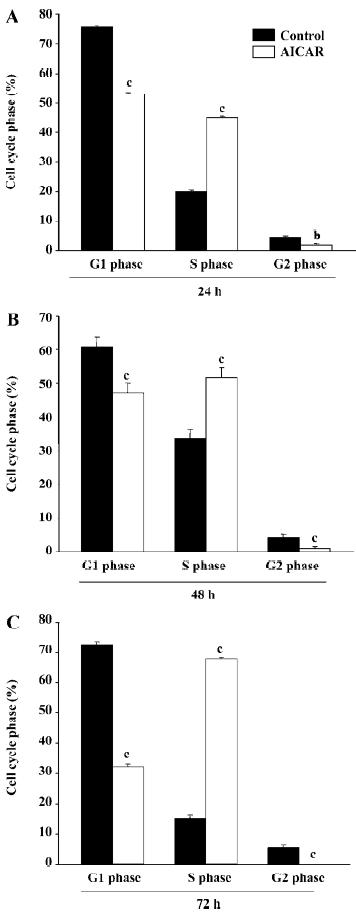
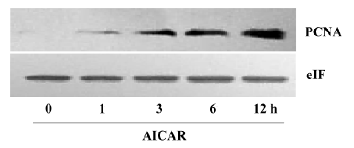
AICAR arrested cell cycle at S-phase The growth of mammalian cells is tightly controlled by cell cycles. To examine whether AICAR affects cell cycles, we stimulated CaSki cells with 500 µmol/L AICAR and analyzed the cell cycles with the flow cytometer. AICAR significantly decreased the number of cells at both the G1 and G2 phases, but increased that of the cells at the S phase when measured at 24, 48, and 72 h (Figure 2A–2C). Therefore, the CaSki cells were arrested at the S phase by AICAR. We also measured the expression of PCNA, an important S-phase marker, by Western blotting. The results showed that the PCNA expression was obviously elevated in a time-dependent manner starting from 1 to 12 h after being normalized with internal control protein eIF5 (Figure 3). This further confirmed that the cells accumulated at the S phase.
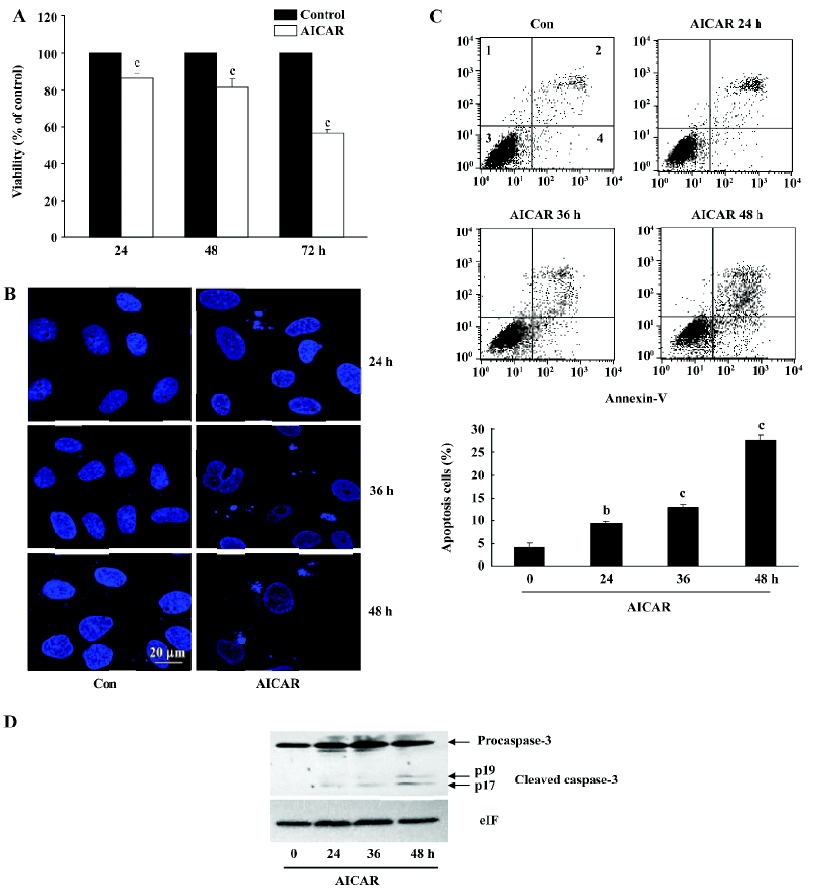
AICAR induces apoptosis in CaSki cells The reduction of the cell number by AICAR in CaSki cells could also be due to the induction of apoptosis. We therefore examined whether AICAR would induce apoptosis by MTT assay, analysis of nuclear morphology, Annexin-V staining, and measurement of caspase-3 activation. As shown in Figure 4A, 500 µmol/L AICAR time-dependently decreased the viability of CaSki cells from 24 to 72 h. The CaSki cells treated with AICAR showed less viable cells compared with the control, and showed more apoptotic cells which appeared as membrane blebbing, cell condensation, chromatin condensation, and formation of an apoptotic body (Figure 4B). One of the key features in apoptotic cells is the translocation of phosphati-dylserine (PS) from the inner to the outer leaflet of the plasma membrane. Annexin-V has a strong affinity for PS and thus can measure apoptosis. As shown in Figure 4C, incubation with AICAR increased the Annexin-positive cells from 4.2%±0.93% to 9.36%±0.41%, 12.78%±0.80%, and 27.59%±1.17% at 24, 36, and 48 h, respectively. Furthermore, the cleaved activated form of caspase-3 was detected and time-dependently increased in the cells treated with AICAR for 24, 36, and 48 h (Figure 4D). Taken together, these results suggested that AICAR not only inhibited proliferation, but also induced apoptosis in CaSki cells.
AICAR suppresses the AKT/mTOR pathway and enhances the p53/ERK pathway
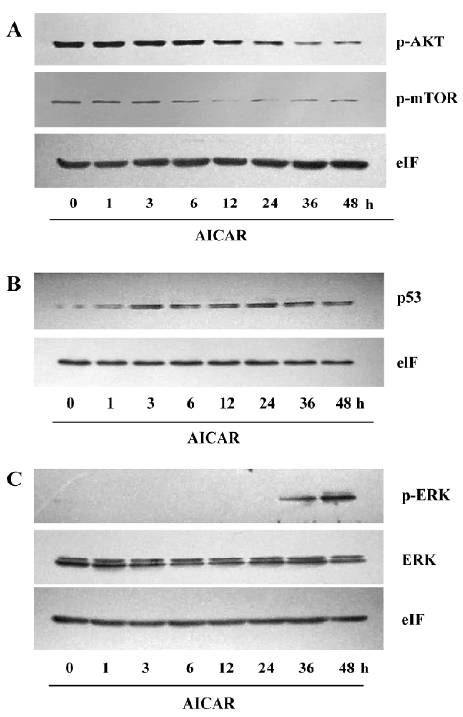
The PI3K/AKT signaling pathway is crucial to many aspects of cell growth, survival, and apoptosis[11]. The constitutive activation of AKT has been involved in the pathogenesis and progression of cervical cancer[12]. Under basal level, AKT was constitutively phosphorylated in CaSki cells (Figure 5A). AICAR did not change the phosphorylation level of AKT when treated for 6 h, but the phosphorylation of AKT was time dependently inhibited by AICAR treatment between 12 and 48 h. We also measured the activation of mTOR, which is one of the important substrates of AKT. It showed a similar pattern of inhibition like AKT. An obvious inhibition of mTOR by AICAR was observed at 12 h (Figure 5A). These results indicated that AICAR may inhibit proliferation and induce apoptosis partially by the repression of the AKT/mTOR pathway. p53 plays a crucial role in the proliferation and survival of cancer cells. At basal level, the p53 protein expressed was quite low, but treatment with AICAR could time-dependently elevate the p53 protein expression following 3 h of stimulation (Figure 5B). We also examined the phosphorylated status of ERK, which was identified as an important mediator of apoptosis in CaSki cells downstream of p53[3]. Our results showed that the phosphorylated ERK at basal level could not be detected in our system and it remained invisible within 12 h after incubation with AICAR. Intriguingly, AICAR increased ERK phosphorylation at 24 h, and further enhanced the phosphorylation at 36 and 48 h (Figure 5C). Taken together, these results demonstrated that AICAR inhibited the AKT/mTOR pathway and potentiated the p53/ERK pathway, which may be responsible for the inhibition of AICAR on proliferation and the induction of apoptosis.
Discussion
In the present study, we found that AICAR inhibited proliferation and induced apoptosis in CaSki cells. This is the first time that it has been demonstrated that AICAR arrests the cell cycle of CaSki cells at the S phase. It was found that AICAR inhibited the AKT/mTOR pathway and upregulated the p53/ERK pathway, which was probably associated with the inhibition of proliferation and the induction of apoptosis by AICAR.
As a cellular energy sensor, AMPK has recently been found to be an important regulator of cell growth, proliferation, and apoptosis[6]. AICAR is an extensively used activator of AMPK. It is transported inside the cells through the adenosine transporter and phosphorylated by adenosine kinase to form zeatin riboside-5-monophosphate, which activates AMPK by mimicking AMP. The activation of AMPK with AICAR significantly inhibited the proliferation of CaSki cells as indicated by the results from the cell count and MTT assay. This observation is consistent with a previous study in other cells[8]. Interestingly, the lower dose of AICAR at a concentration of 100 µmol/L showed an obvious inhibition of proliferation at 24 h, but had no effect at 48 and 72 h. AICAR at 250 µmol/L could significantly reduce the cell number at 24 and 72 h but not at 48 h. This may indicate that cervical cancer cells exhibit different sensitivity to AMPK activation at different times, and it is also possible that these doses induced an approximately equivalent counteracting force of proliferation in the cells, resulting in the fluctuation of cell numbers. Further studies are still needed to elucidate this paradox. Our results demonstrated that 500 µmol/L AICAR had already exerted a strong suppression on pro-liferation, and this concentration was much lower than that used in other cells which ranged from 1 and 5 mmol/L[13]. Cell proliferation is regulated by cycle machinery, which are balanced by cyclin-dependent kinases (CDK), CDKinhibitors (CDKI), and tumor suppressors[6]. In some cancer cells, AICAR could upregulate the expression of CDKI, like P21 and p27, and elevate the level of p53, leading to cell cycle arrest at the G1 phase[8]. Interestingly, the effect of AICAR on CaSki cells is quite different. AICAR decreased the number of cells at the G1 and G2 phases and increased the number of cells at the S phase, resulting in an attenuation of cells entering the G2 phase (Figure 2). Thus, PCNA, an index of the S phase, was also upregulated by AICAR (Figure 3). The entry of the G2 phase is mainly regulated by cyclin A and cyclin B[14], and it remains unknown as to whether AICAR affects these 2 cyclins.
The reduction of the cell number may also be due to apoptosis or cell death. The results of the cell viability showed an obvious decrease of living cells in the AICAR-treated group (Figure 4A). CaSki cells with AICAR stimulation also had typical morphological changes of apoptosis (Figure 4B). Furthermore, AICAR increased Annexin-V positive cells and activated caspase-3, a key effector of the apop-totic signaling pathways (Figure 4C, 4D). These results indicated that AICAR not only inhibited proliferation, but induced apoptosis in cervical cancer cells as well. The PI3K pathway plays an important role in cervical cancer[4]. As a cardinal downstream effector, AKT was time dependently dephosphorylated by AICAR (Figure 5A). The effect of AICAR on AKT phosphorylation varies for different cell types. In vessel smooth muscle cells[15] and lung cancer cells[16], there was no change of AKT phosphorylation by AICAR, while AICAR could inhibit AKT phosphorylation in several cancer cell lines[8]. mTOR is an important substrate of AKT and we found that AICAR inhibited mTOR phosphorylation (Figure 5A), which further supports the suppressive effect of AICAR on the PI3K pathway. Rapamycin, a mTOR inhibitor, now has been used clinically as an anticancer drug[17]. Our observation suggests that AICAR may be an important potential drug in cervical cancer treatment.
Most cervical cancer cases are found to be infected with HPV, and the viral E6 protein forms a ternary complex with the cellular E6–AP protein, which expedites the degradation of p53 in these cells. Therefore, p53 expression is relative lower in cervical cancer cells. We found AICAR could upregu-late p53 expression (Figure 5B). This result is consistent with some previous reports in other cell types[8]. As an important tumor suppressor, p53 could regulate CDK, CDKI, and many other substrates to regulate cell growth. A recent study suggested that ERK activation were also important in DNA damage-induced apoptosis[18]. Singh et al[3] reported that in SiHa and CaSki cells ERK activation was required in carboplatin-induced apoptosis, and p53 regulated the activation of ERK. In agreement with this new pathway, our results showed that AICAR apparently elevated the phosphorylation of ERK (Figure 5C).
In conclusion, we found that AICAR inhibited proliferation and induced apoptosis in cervical cancer cells, which suggests that AMPK may be an important potential target for cervical cancer treatment and AICAR may be a useful therapeutic drug against cervical cancer.
References
- Au WW, Abdou-Salama S, Al-Hendy A. Inhibition of growth of cervical cancer cells using a dominant negative estrogen receptor gene. Gynecol Oncol 2007;104:276-80.
- Lee JJ, Kim S, Yeom YI, Heo DS. Enhanced specificity of the p53 family proteins-based adenoviral gene therapy in uterine cervical cancer cells with E2F1-responsive promoters. Cancer Biol Ther 2006;5:1502-10.
- Singh S, Upadhyay AK, Ajay AK, Bhat MK. p53 regulates ERK activation in carboplatin induced apoptosis in cervical carcinoma: a novel target of p53 in apoptosis. FEBS Lett 2007;581:289-95.
- Lee CM, Fuhrman CB, Planelles V, Peltier MR, Gaffney DK, Soisson AP, et al. Phosphatidylinositol 3-kinase inhibition by LY294002 radiosensitizes human cervical cancer cell lines. Clin Cancer Res 2006;12:250-6.
- Faried LS, Faried A, Kanuma T, Nakazato T, Tamura T, Kuwano H. Inhibition of the mammalian target of rapamycin (mTOR) by rapamycin increases chemosensitivity of CaSki cells to paclitaxel. Eur J Cancer 2006;42:934-47.
- Motoshima H, Goldstein BJ, Igata M, Araki E. AMPK and cell proliferation-AMPK as a therapeutic target for atherosclerosis and cancer. J Physiol 2006;574:63-71.
- Luo Z, Saha AK, Xiang X, Ruderman NB. AMPK, the metabolic syndrome and cancer. Trends Pharmacol Sci 2005;26:69-76.
- Rattan R, Giri S, Singh AK, Singh I. 5-Aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside inhibits cancer cell proliferation in vitro and in vivo via AMP-activated protein kinase. J Biol Chem 2005;280:39582-93.
- Xiang X, Saha AK, Wen R, Ruderman NB, Luo Z. AMP-activated protein kinase activators can inhibit the growth of prostate cancer cells by multiple mechanisms. Biochem Biophys Res Commun 2004;321:161-7.
- Kimball SR. Interaction between the AMP-activated protein kinase and mTOR signaling pathways. Med Sci Sports Exerc 2006;38:1958-64.
- Martelli AM, Nyakern M, Tabellini G, Bortul R, Tazzari PL, Evangelisti C, et al. Phosphoinositide 3-kinase/Akt signaling pathway and its therapeutical implications for human acute myeloid leukemia. Leukemia 2006;20:911-28.
- Hagemann T, Bozanovic T, Hooper S, Ljubic A, Slettenaar VI, Wilson JL, et al. Molecular profiling of cervical cancer progres-sion. Br J Cancer 2007;96:321-8.
- Nafz J, De-Castro Arce J, Fleig V, Patzelt A, Mazurek S, Rosl F. Interference with energy metabolism by 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside induces HPV suppression in cervical carcinoma cells and apoptosis in the absence of LKB1. Biochem J 2007;403:501-10.
- Park MT, Lee SJ. Cell cycle and cancer. J Biochem Mol Biol 2003;36:60-5.
- Nagata D, Takeda R, Sata M, Satonaka H, Suzuki E, Nagano T, et al. AMP-activated protein kinase inhibits angiotensin II-stimulated vascular smooth muscle cell proliferation. Circulation 2004;110:444-51.
- Carretero J, Medina PP, Blanco R, Smit L, Tang M, Roncador G, et al. Dysfunctional AMPK activity, signalling through mTOR and survival in response to energetic stress in LKB1-deficient lung cancer. Oncogene 2007;26:1616-25.
- Mita MM, Mita A, Rowinsky EK. The molecular target of rapamycin (mTOR) as a therapeutic target against cancer. Cancer Biol Ther 2003;2 Suppl 1:S169-77.
- Tang D, Wu D, Hirao A, Lahti JM, Liu L, Mazza B, et al. ERK activation mediates cell cycle arrest and apoptosis after DNA damage independently of p53. J Biol Chem 2002;277:12710-7.