
Pseudolaric acid B induces apoptosis, senescence, and mitotic arrest in human breast cancer MCF-7
Introduction
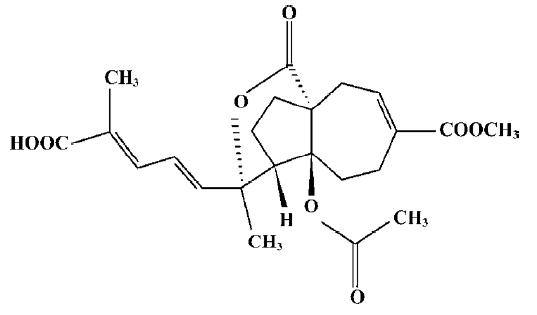
Pseudolaric acid B (PAB; Figure 1) is a diterpene acid isolated from the root and trunk bark of Pseudolarix kaempferi Gordon (Pinaceae), known as “Tu-Jin-Pi” in Chinese, which has been used to treat dermatological fungi infections. PAB exerts potent antifungal, antimicrobial[1], antifertility[2,3], and cytotoxic activity in vitro[4], and the mechanisms of PAB-induced cell death in human myelocytic leukemia K562, human cervical carcinoma HeLa, and human melanoma A375-S2 in vitro have been reported[5–7]. However, the mechanism of PAB-induced cell death in human breast cancer MCF-7 is not clear. In the present study, PAB is shown to inhibit MCF-7 cell proliferation through apoptosis and senescence.
Complex organisms have evolved at least 2 cellular mechanisms to suppress cell proliferation at risk of oncogenic transformation: apoptosis and cellular senescence in particular. Apoptosis kills and eliminates potential cancer cells, and cellular senescence irreversibly arrests their growth[8]. Some chemicals, such as doxorubicin, simultaneously induce apoptosis, senescence, and necrosis in a concentration-dependent manner. Caspase-3 is mainly responsible for apoptosis, while p53 for senescence. It has been reported that the inhibition of caspases can switch doxorubicin-induced apoptosis to senescence[9]. MCF-7 cells are characterized by wild-type p53 and defective caspase-3 functions, therefore, they were selected as the senescence model.
At key transitions during eukaryotic cell cycle progression, signaling pathways monitor the successful completion of upstream events before proceeding to the next phase. These regulatory pathways are commonly referred to as cell cycle checkpoints[10]. Cells are arrested at cell cycle checkpoints temporarily to allow for: (i) cellular damage to be repaired; (ii) the dissipation of an exogenous cellular stress signal; or (iii) the availability of essential growth factors, hormones, or nutrients. Checkpoint signaling may also result in the activation of pathways leading to programmed cell death, if cellular damage can not be properly repaired[11]. Cell cycle arrest is necessary to senescence, because the experimental inactivation of DNA-damage checkpoint response abrogates oncogene-induced senescence. DNA-damage checkpoint response is triggered by oncogene-induced DNA hyper-replication[12], hence cell cycle arrest should happen after the S phase.
Cyclin/Cdk complexes play a key role in the cell cycle progression. Cdk is negatively regulated by a group of functionally-related proteins called Cdk inhibitors. p21Waf1/Cip1 belongs to Cip/Kip inhibitors, and the Cip/Kip family takes charge of all phases of the cell cycle[13]. Wild-type p53 is involved in essential functions, such as DNA repair, transcription, genomic stability, senescence, cell cycle control, and apoptosis. A major target gene which participates in p53-mediated growth arrest is p21Waf1/Cip1[14]. It is reported that in cell cycle progression, cyclin B1 (whose levels rise during the S and G2 phases) then was transported to nuclei, and peaks in mitosis[15]. Cyclin B1 in nuclei must be destroyed before the cells escape from mitosis[16]. Therefore, the expression of p21, p53, and cyclin B1 was investigated in this study.
Materials and methods
Materials PAB, which was purchased from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China), was dissolved in DMSO to make a stock solution. The DMSO concentration was kept below 0.01% in all the cell cultures, and did not exert any detectable effect on cell growth or cell death. Propidium iodide (PI), RNase A, proteinase K, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), DMF (dimethylforma-mide), and acridine orange were purchased from Sigma (St Louis, MO, USA). The senescence detection kit was purchased from Calbiochem (La Jolla, CA, USA). Rabbit polyclonal antibodies against cyclin B1, p21, p53, caspase-3, Fas-associated death domain FADD, Fas L, β-actin, and horseradish peroxidase-conjugated secondary antibodies (goat-antirabbit) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Agonistic anti-Fas immunoglobulin M mAb (clone CH-11) and anti-Fas immunoglobulin G (clone UB2) were purchased from Medical and Biological Laboratories (Nagoya, Japan)
Cell culture Human breast cancer MCF-7 cells were obtained from American Type Culture Collection (Manassas, VA, USA) and were cultured in RPMI-1640 medium (Hyclone, Logan, UT, USA) supplemented with 10% heat-inactivated (56 °C, 30 min) fetal calf serum (Beijing Yuanheng Shengma Research Institution of Biotechnology, Beijing, China), 2 mmol/L glutamine (Gibco, Grand Island, NY, USA), penicillin (100 U/mL), and streptomycin (100 µg/mL), and maintained at 37 °C with 5% CO2 in a humidified atmosphere.
Cell growth inhibition test The inhibition of cell growth was determined by a MTT test. The MCF-7 cells (1.5×104 cells/well) were seeded into 96-well culture plates (Nunc, Roskilde, Denmark). After overnight incubation, various concentrations of PAB, CH11, or UB2 were added to the plates. Following incubation, cell growth was measured at different time points by the addition of MTT at 37 °C for 3 h; DMSO (150 µL) was added to dissolve the formazan crystals. Absorbance was measured at 492 nm with an ELISA plate reader (Bio-Rad, Hercules, CA, USA). The percentage of inhibition was calculated as follows:
Cell death (%)=(A492 [control]-A492 [sample])/
A492 [control]×100%.
Observation of morphological changes by light microscopy The MCF-7 cells were treated with PAB (0 and 4 µmol/L) for 36 h. The morphological changes were observed by phase contrast microscopy (Leica, Nusslich, Germany).
Nuclei alternation observed by acridine orange staining The MCF-7 cells, which were incubated in RPMI-1640 containing 10% fetal calf serum, were seeded into 6-well plates (Nunc, Denmark) with coverslips and cultured overnight. The cells were treated with 0 and 4 µmol/L PAB for 36 h. The morphological changes of the nuclei were observed by acridine orange staining. The cells on the coverslips were rinsed and stained with acridine orange (10 mg/L) at 37 °C for 30 min. After the coverslips were sealed, the samples were observed by fluorescence microscopy (Leica, Germany).
Determination of DNA fragmentation by agarose gel electrophoresis[17] The MCF-7 cells were treated with 0, 1, 2, 4, and 10 µmol/L PAB for 36 h, and then both the adherent and floating cells were collected by centrifugation at 1000×g for 5 min. The cell pellet was suspended in cell lysis buffer [10 mmol/L Tris-HCl (pH 7.4), 10 mmol/L EDTA acid (pH 8.0), and 0.5%Triton-100], and kept at 4 °C for 30 min. The lysate was centrifuged at 25 000×g for 20 min. The supernatant was incubated with 20 g/L RNase A (2 µL) at 37 °C for 1 h, then incubated with 20 g/L proteinase K (2 µL) at 37 °C for 1 h. The supernatant was mixed with 5 mol/L NaCl (20 µL) and isopropanol (120 µL) at -20 °C over-night, and then centrifuged at 25 000×g for 15 min. After discarding the supernatant, the DNA sediment was dissolved in 20 µL TE buffer [10 mmol/L Tris-HCl (pH 7.4) and 1 mmol/L EDTA (pH 8.0)], and separated by 2% agarose gel electrophoresis at 100 V for 50 min.
Lactate dehydrogenase activity-based cytotoxicity assays[18] The cells were cultured with 4 µmol/L PAB for 12, 24, 36, and 48 h. Floating dead cells were collected from the culture medium by centrifugation (240×g for 10 min at 4 °C), and the lactate dehydrogenase (LDH) content from the pellets lysed in 0.1% Nonidet P 40 (NP-40) for 15 min was used as an index of apoptotic cell death (LDHp). The LDH released into the culture medium [extracellular LDH (LDHe)] was used as an index of necrotic cell death. The adherent and viable cells were lysed in 0.1% NP-40 for 15 min to release LDH [intracellular LDH (LDHi)]. The substrate reaction buffer of 0.5 mmol/L LDH (L [+]-lactic acid, 0.66 mmol/L indonitrotetrazolium, 0.28 mmol/L phenazine methosulfate, 1.3 mmol/L nicotinamide adenine dinucleotide in pH 8.2 Tris-HCl) was added. The A value at 490 nm of the reaction for 1 and 5 min was assayed and the formula was as follows:
LDH activity=(A5 min–A1 min)/4.
The percentage of apoptotic and necrotic cell death was calculated as follows:
Apoptosis (%)=LDHp/(LDHp+LDHe+LDHi)×100;
Necrosis (%)=LDHe/(LDHp+LDHe+LDHi)×100.
SA-β-galactosidase detection[19] The MCF-7 cells (1.5×105 cells/well) were seeded into 24-well culture plates (Nunc, Denmark). After overnight incubation, 4 µmol/L PAB was added to the plates. After 3 d of incubation, the surviving MCF-7 cells were cultured for 5 d in fresh medium. Following the protocol of the senescence detection kit, the culture medium were removed and the cells were washed once with 1 mL phosphate-buffered saline (PBS), then the cells were fixed with 0.5 mL of fixative solution at room temperature for 10–15 min. The staining solution mix was prepared in a polypropylene plastic tube. For each well, 470 µL staining solution, 5 µL staining supplement, and 25 µL 20 mg/mL X-gal in DMF were added. The cells were rinsed twice with 1 mL PBS; 0.5 mL of the staining solution mix was added to each well and then incubated at 37 °C without CO2 overnight. The cells were observed under a microscope for the development of a blue color, and the number of blue-stained cells in 100 cells was calculated.
Flow cytometric analysis[20] The MCF-7 cells were harvested and rinsed with PBS. The cell pellets were fixed in 70% ethanol at 4 °C overnight. After washing twice with PBS, the cells were stained with 1.0 mL PI solution containing 50 mg/L PI, 1 g/L RNase A, and 0.1% Triton X-100 in 3.8 mmol/L sodium citrate, followed by incubation on ice in the dark for 30 min. The samples were analyzed by a FACScan flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA).
Western blot analysis of protein expression[21] The MCF-7 cells were treated with 4 µmol/L PAB for the indicated times. Both adherent and floating cells were collected and frozen at -80 °C. A Western blot analysis was performed for the total proteins as follows. Briefly, the cell pellets were resuspended in lysis buffer, including 50 mmol/L 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (pH 7.4), 1% Triton-X 100, 2 mmol/L sodium orthovanadate, 100 mmol/L sodium fluoride, 1 mmol/L EDTA, 1 mmol/L egtazic acid (EGTA), 1 mmol/L phenyl-methylsulfonyl fluoride (PMSF), 0.1 g/L aprotinin, and 0.01 g/L leupeptin, then lysed at 4 °C for 1 h. After 13 000×g centrifugation for 10 min, the protein content of the supernatant was determined using Bio-Rad protein assay reagent (Bio-Rad, USA). The protein was loaded in each lane, then separated by 12% SDS–PAGE, and blotted onto a nitrocellulose membrane. The protein expression was detected using primary polyclonal antibody (1:200–1000) and secondary polyclonal antibody (1:500) conjugated with peroxidase. For the cytoplasmic and nuclear proteins, the protein content was determined as follows: the cell pellets were resuspended in 60 µL lysis buffer, including 20 mmol/L HEPES, 10 mmol/L KCl, 1.5 mmol/L MgCl2, 1 mmol/L EDTA, 1 mmol/L EGTA, 1 mmol/L dithiothreitol (DTT), and 1 mmol/L PMSF (pH 7.5), then lysed on ice at 4 oC for 1 h. After 16 000×g centrifugation for 20 min, the supernatant for the cytoplasm proteins was collected. The pellet was resuspended in 15 µL lysis buffer, including 20 mmol/L HEPES, 25% glycerol, 420 mmol/L NaCl, 1.5 mmol/L MgCl2, 0.2 mmol/L EDTA, 0.5 mmol/L DTT, 0.5 mmol/L PMSF, and 5 mg/L leupeptin, and lysed at 4 °C for 15 min on ice. After 12 000×g centrifugation for 10 min, the nuclear proteins were contained in the supernatant.
Statistical analysis All data represent at least 3 independent experiments and are expressed as mean±SD. Statistical comparisons were made using Student’s t-test. P-values of less than 0.05 represented a statistically significant difference.
Results
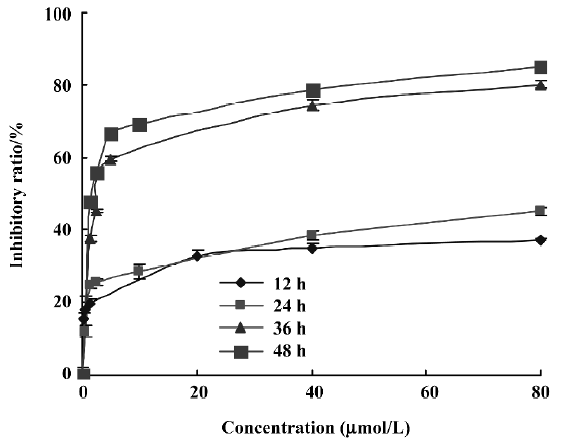
Cytotoxic effect of PAB on cell growth To detect the growth inhibition of PAB-exposed MCF-7cells, the cells were treated with various doses of PAB, ranging from 0.6 to 80 µmol/L for 12, 24, 36, and 48 h, and the half maximal inhibitory concentration IC50 values were 682.2, 165.8, 3.4, and 1.35 µmol/L at 12, 24, 36, and 48 h, respectively. PAB showed a slow but potent suppressive effect on the MCF-7 cells. The cytotoxicity was increased in a time- and dose-dependent manner (Figure 2). In the following experiments, we adopted 4 µmol/L at 36 h as the best concentration. At 12 and 24 h, the death ratio was low after 4 µmol/L PAB treatment.
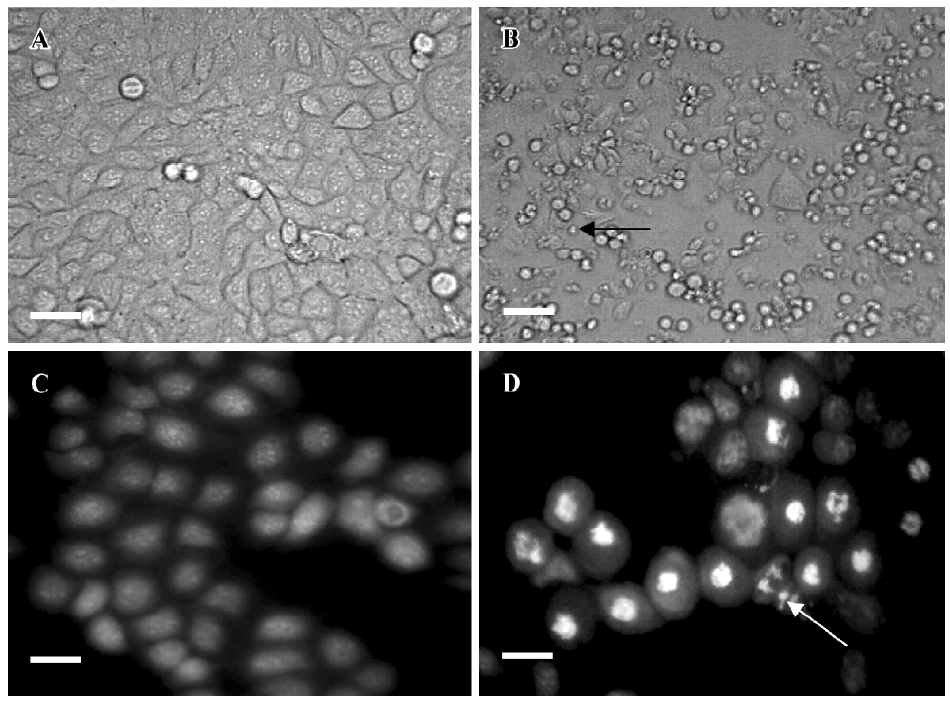
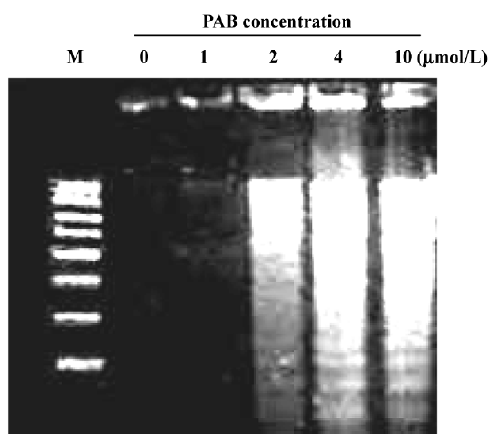
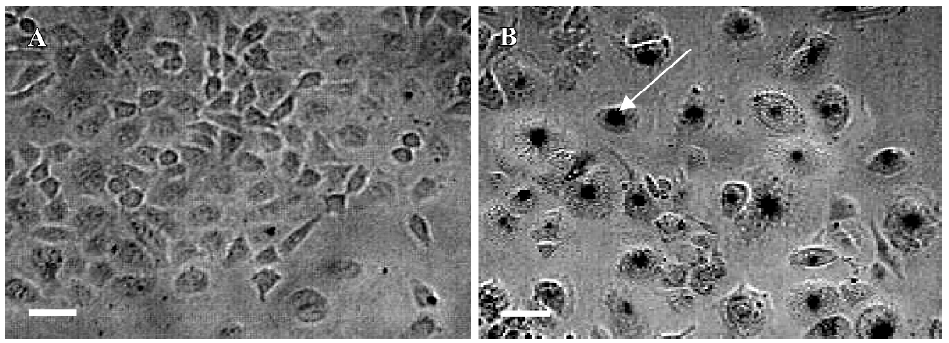
Effects of PAB on morphological changes and DNA fragmentation Morphological changes were observed by phase contrast microscopy and fluorescence microscopy. After 36 h with 4 µmol/L PAB treatment, we observed a decrease in the total cell number, an increase in floating cells, and the appearance of apoptotic bodies (Figure 3B). Nuclear changes were also detected by acridine orange staining. In the control group, the MCF-7 cells were intact in shape and stained green homogeneously (Figure 3C). After 36 h with PAB 4 µmol/L treatment, bright green nuclei blebbing and DNA fragmentation were observed (Figure 3D). DNA fragmentation became obvious after 4 and 10 µmol/L PAB treatments for 36 h on agarose gel electrophoresis (Figure 4). Taken together, the major cause of cell death induced by PAB was through the apoptotic pathway.
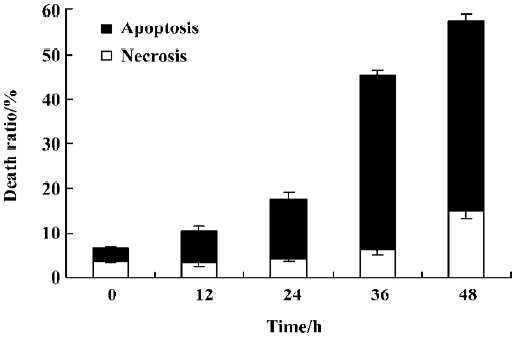
Characteristics of cell death identified by the LDH release assay It was reported that the rate of LDH released from viable cells, floating dead cells, and the culture medium was used to distinguish the proportion of apoptotic and necrotic cells[17]. The number of apoptotic MCF-7 cells increased from 3% at 0 h to 39% at 36 h in the presence of 4 µmol/L PAB. However, the percentage of necrotic cells was still negligible (Figure 5). These results were consistent with morphological changes and DNA fragmentation (Figure 4), suggesting that the major cause of PAB-treated MCF-7 cell death was apoptosis at 36 h. At 12 h, the apoptotic ratio was very low after 4 µmol/L PAB treatment.
Detection of senescence Mammalian cells express lysosomal β-galactosidase activity, which is measured at pH 6.0. SA-β-galactosidase has become widely accepted as an important biomarker for senescence[19]. MCF-7 cells were cultured for 5 d in fresh medium after 3 d of incubation with 4 µmol/L PAB, then the surviving cells were detected. In total, 96% of the cells were positive for SA-β-galactosidase blue staining. Cell morphology showed gross enlargement, flattening, and accumulation of granular cytoplasmic inclusion (Figure 6).
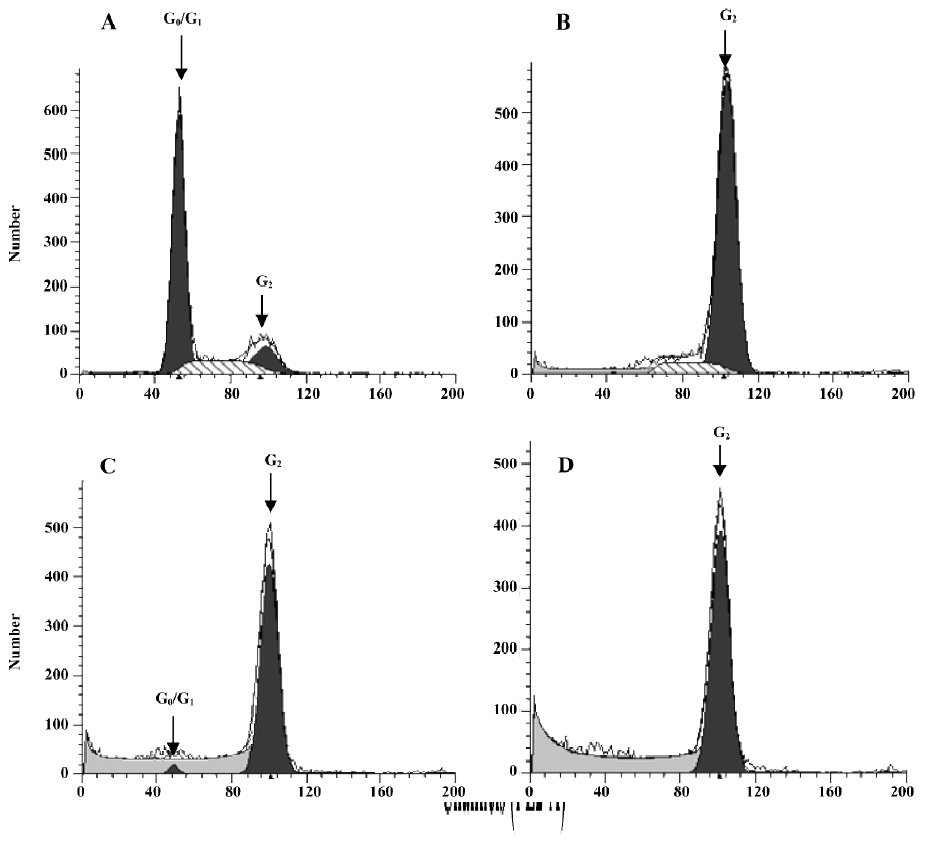
Effect of PAB on the cell cycle To further investigate the mechanism of cell growth inhibition by PAB, a flow cytometric analysis was performed. After 4 µmol/L PAB treatment for 0, 12, 24, and 36 h, the DNA amount was doubled compared with the control group (Figure 7), indicating the PAB-treated cells might be arrested at the G2 or M phases. The samples were analyzed by Cell Quest software (Becton Dickinson, USA), which determined the percentage of cells at different phases of the cell cycle.
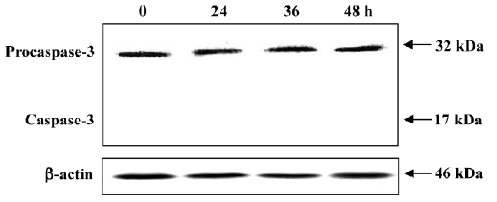
No procaspase-3 degradation in PAB-induced MCF-7 cell death MCF-7 cells express wild-type p53, but are absent of the functional caspase-3 protein[22]. As shown in Figure 8, there was constant expression of non-functional procaspase-3 after 4 µmol/L PAB treatment at 0, 24, 36, and 48 h, but no expression of active caspase-3 (17 kDa). At different time points, the expression of procaspase-3 was not altered, indicating that PAB exerted no effect on caspase-3 activity (Figure 8).
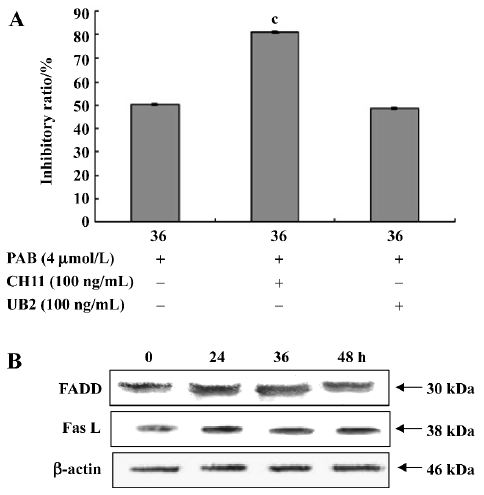
Apoptosis induced by PAB was independent of death receptor pathways Anti-Fas agonistic antibody CH11 activated the Fas receptor resulting in apoptosis, but anti-Fas antagonistic antibody UB2 can block this pathway and prevent cell death. At 36 h, the inhibitory ratio of the PAB group was 50.4%, the CH11+PAB group was 80.9%, and the UB2+PAB group was 48.7% (Figure 9A). The result from the Western blot analysis showed that the expression of FADD and Fas L was stable at different time points (Figure 9B).
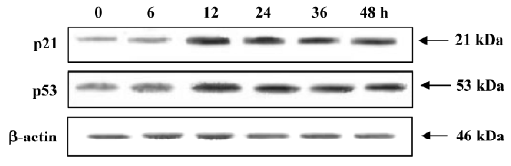
Involvement of p21 and p53 in the cycle arrest of MCF-7 cells When exposed to 4 µmol/L PAB, the expression of p21 and p53 was upregulated from 12 h and the expression levels were stable for a further 12 h (Figure 10).
Translocation of cyclin B1 participation in mitotic arrest of MCF-7 cells after PAB treatment After PAB treatment, the amount of DNA was doubled, but the arrested phase, G2 or M, was not confirmed. Then, the cyclin B1 expression was compared in the cytoplasm and nuclei. After PAB treatment, total cyclin B1 was increased from 12 to 36 h, but after 48 h it began to decrease. The expression of cyclin B1 in nuclei was increased at 12 and 24 h, and then remained stable. However, its expression in the cytoplasm was significantly upregulated at 12 h, and then decreased at 24 h (Figure 11).

Discussion
Apoptosis and senescence, which have different characteristics, exert anticancer effects. As a mechanism of cell death, apoptosis is an important process for normal development and suppression of oncogenesis. Apoptosis is characterized by a series of typical morphological events, such as cell shrinkage, DNA fragmentation, and fragmentation into membrane-bound apoptotic bodies and rapid phagocytosis by neighboring cells[24]. PAB obviously inhibited MCF-7 cell growth in a time- and dose-dependent manner, and had a potent suppressive effect on the MCF-7 cells until 36 h treatment. The appearance of apoptotic bodies and DNA fragmentation were observed after 4 µmol/L PAB treatment. Apoptotic bodies and DNA fragmentation are regarded as the apoptotic hallmarks. The LDH activity-based cytotoxicity assay also demonstrated that in MCF-7 cells, the apoptotic ratio was dominant at 36 h after 4 µmol/L PAB treatment. Therefore, PAB induced the death of MCF-7 cells mainly through apoptosis. Senescent cells displayed morphological features, such as enlargement, flattening, and positive SA-β-galactosidase staining. The surviving MCF-7 cells, which were cultured for 5 d in fresh medium after 3 d of incubation with 4 µmol/L PAB, possessed these typical senescent phenotype. Therefore, senescence was also induced by the same concentration of PAB in a different manner compared with apoptosis in the MCF-7 cells.
Apoptotic processes led to the activation of caspase-3 in a variety of cells which induced proteolytic cleavage of its substrates, including the DNA fragmentation. MCF-7 cells with wild-type p53, are absent of caspase-3 activity; however, inactive procaspase-3 was expressed in the MCF-7 cells. The appearance of the active form of caspase-3 was not observed, and the expression of procaspase-3 was unchanged with the time-course in MCF-7 cells at our laboratory. The result is consistent with that of Cui et al[25]. The appearance of procaspase-3 may be the result of the mutation in MCF-7 cells, but MCF-7 cells in this study did not have functional caspase-3. It is reported that DNA fragmentation independent of caspase-3 is induced by some factors, such as apoptosis-inducing factor AIF[26], therefore in this study, the appearance of the DNA ladder is independent of caspase-3, but the detailed mechanism remains to be elucidated. In apoptosis, after the binding of death ligands, such as Fas L or agonistic antibodies to their cognate receptors, a cytosolic adaptor protein FADD is recruited to the cytoplasmic domain of the Fas to trigger the death receptor pathway[27]. In this study, the CH11 agonistic Fas antibody induced the increasing death of PAB-treated cells, but the UB2 antagonistic antibody-treated group had no such effect on the death ratio, indicating that there was another pathway, but not death receptor pathway, to take effect in the apoptosis of MCF-7 cells. The Western blotting analysis further indicated that the stable expression of FADD and Fas L was not involved in apoptosis. The detailed apoptotic pathway induced by PAB remains to be elucidated.
Cell cycle arrest may be attributed to the activation of pathways leading to programmed cell death and senescence. Cycle arrest contributes to apoptosis[11], but only cell cycle arrest which happens after the S phase is in charge of senescence[11,19]. In this study, PAB induced obvious mitotic arrest, which was preceded by apoptosis and senescence in time, suggesting that PAB-induced apoptosis and senescence might be caused by mitotic arrest in MCF-7 cells. It has been reported that p21 is responsible for all cell cycle arrest[13]. PAB 4 µmol/L treatment for 12 h could activate p21. Since p53 resides at the upstream of p21[28-30], and 4 µmol/L PAB treatment for 12 h could induce the expression of p53, PAB might increase the expression of p21 by upregulating the expression of p53. It has been reported that cyclin B1 increases in mitosis[16,23]. In the present study, 4 µmol/L PAB increased the expression of total cyclin B1. It is found that cyclin B1 transports into nuclei in mitosis, and in nuclei it must be destroyed before cells escape from mitosis[16]. After 4 µmol/L PAB treatment for 12 h, nuclear cyclin B1 was increased and not degraded. The cytoskeleton mainly consists of microfilaments and microtubules that form a dynamic framework that maintains the cell shape[31]. Microtubules are highly dynamic polymers that switch between phases of extension and shortening at individual microtubule ends[32]. This process, known as microtubule dynamics, is particularly important for mitosis when the rapid reorganization of microtubules is required for the alignment of chromosomes, as well as for subsequent chromosome separation[33,34]. One of the key characteristics of antimicrotubule agents is the induction of mitotic arrest of the cell cycle[35]. As a microtubule-disrupting agent, it is reported that PAB exerts its effect by directly interacting with tubulin, disrupting the formation of mitotic spindles and microtubules[36,37]. Taken together, these data showed that PAB treatment did not activate the G2 checkpoint, and suggested that PAB-treated cells were probably arrested at this mitotic phase of the cell cycle.
Taken together, we conclude that PAB could induce mitotic arrest, apoptosis, and senescence in MCF-7 cells. PAB induced apoptosis through a pathway other than death receptor pathway. Mitotic arrest was associated with the upregulation of p21, p53, and cyclin B1. PAB promoted the transportation of cyclin B1 from the cytoplasm to nuclei, and inhibited cyclin B1 degradation in nuclei.
References
- Li E, Clark AM, Hufford CD. Antifungal evaluation of PAB, a major constituent of Pseudolarix kaempferi. J Nat Prod 1995;58:57-67.
- Wang WC, Lu RF, Zhao SX, Gu ZP. Comparison of early pregnancy-terminating effect and toxicity between pseudolaric acids A and B. Acta Pharmacol Sin 1988;9:445-8.
- Wang WC, Lu RF, Zhao SX, Zhu YZ. Antifertility effect of PAB. Acta Pharmacol Sin 1982;3:188-92.
- Pan DJ, Li ZL, Hu CQ, Chen K, Chang JJ, Lee KH. The cytotoxic principles of Pseudolarix Kaempferi: Pseudolaric acid-A and B and related derivatives. Planta Med 1990;56:383-5.
- Gong XF, Wang MW, Tashiro SI, Onodera S, Ikejima T. PAB induces apoptosis through P53 and Bax/Bcl-2 pathways in human melanoma A375-S2 cell. Arch Pham Res 2005;28:68-72.
- Zhang M, Mo X, Chen XY, Chen L, Li LZ. Studies of apoptosis on K562 cell line induced by pseudolaricd acid B in vitro. Chin Tradit Herb Drugs 2002;33:533-5.
- Gong XF, Wang MW, Tashiro SI, Onodera S, Ikejima T. Involvement of JNK-initiated p53 accumulation and phosphorylation of p53 in pseudolaric acid B induced cell death. Exp Mol Med 2006;38:428-34.
- Campisi J. Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol 2001;11:S27-31.
- Rebbaa A, Zheng X, Chou PM, Mirkin BL. Caspase inhibition switches doxorubicin-induced apoptosis to senescence. Oncogene 2003;22:2805-11.
- Hartwell LH, Weinert TA. Checkpoints: controls that ensure the order of cell cycle events. Science 1989;246:629-34.
- Pietenpol JA, Stewart ZA. Cell cycle checkpoint signaling: cell cycle arrest versus apoptosis. Toxicology 2002;181–182:475-81.
- Micco RD, Fumagalli M. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006;444:638-42.
- Sherr CJ, Roberts JM. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev 1995;9:1149-63.
- Caroline CR, Philippe R, Elisheva YR. Mechanisms of P53-induced apoptosis: in search of genes which are regulated during P53-mediated cell death. Toxicol Lett 1998;102–103:491-6.
- Peng CY, Graves PR, Thoma RS, Wu ZQ, Shaw AS, Piwnica-Worms H. Mitotic and G2 checkpoint control: regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science 1997;277:1501-5.
- Murray AW, Kirschner MW. Dominoes and clocks: the union of two views of cell cycle regulation. Science 1989;246:614-21.
- Sarin A, Hadid E, Henkart PA. Caspase dependence of target cell damage induced by cytotoxic T lymphocytes. J Immunol 1998;161:2810-3.
- Kim YM, Talanian RV, Billiar TR. Nitric oxide inhibits apoptosis by preventing increases on caspase-3-like activity via two distinct mechanisms. J Biol Chem 1997; 272: 31 138–47.
- Dimri GP, Leet X, Basile G, Acost M, Scorrt G, Campisi J, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Sci Acad USA 1995;92:9363-7.
- Lee KY, Park JA, Chung E, Lee YH, Kim SI, Lee SK. Ginsenoside Rh2 blocks the cell cycle of SK-KEP-1cells at the G1/S boundary by selectively inducing the protein expression of p27kipl. Cancer Lett 1996;110:193-200.
- Nalini K, Rayudu G, Vsha G, Jinah C, Henry JF. Role of protein kinase C in basal and hydrogen peroxide stimulated NF-κB activation in the murine macrophage. Arch Biochem Biochem Biophys 1998;350:79-86.
- Janicke RU, Sprengart ML, Wati MR, Porter AG. Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J Biol Chem 1998;273:9357-60.
- Jin P, Hardy S, Morgan DO. Nuclear localization of cyclin B1 controls mitotic entry after DNA damage. J Cell Biol 1998;141:875-85.
- Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 1972;26:239-57.
- Cui Q, Yu JH, Wu JN, Tashiro S, Onodera S, Minami M, et al. P53-mediated cell cycle arrest and apoptosis through a caspase-3-independent, but caspase-9-dependent pathway in oridonin-treated MCF-7 human breast cancer cells. Acta Pharmacol Sin 2007;28:1057-66.
- Cregan SP, Dawson VL, Slack RS. Role of AIF in caspase-dependent and caspase-independent cell death. Oncogene 2004;23:2785-96.
- Chinnaiyan AM, O’Rourke K, Tewari M, Dixit VM. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell 1995;81:505-12.
- May P, May E. Twenty years of p53 research: structural and functional aspects of the p53 protein. Oncogene 1999;18:7621-36.
- Macleod KF, Sherry N, Hannon G, Beach D, Tokino T, Kinzler K, et al. P53-dependent and independent expression of p21 during cell growth, differentiation, and DNA damage. Genes Dev 1995;9:935-44.
- Flatt PM, Tang LJ, Scatena CD, Szak ST, Pietenpol JA. P53 regulation of G2 checkpoint is retinoblastoma protein dependent. Mol Cell Biol 2000;20:4210-23.
- Ingber DE, Prusty D, Sun Z, Betensky H, Wang N. Cell shape, cytoskeletal mechanics and cell cycle control in angiogenesis. J Biomech 1995;28:1471-84.
- Mitchison T, Kirschner M. Dynamic instability of microtubule growth. Nature 1984;312:237-42.
- Rusan NM, Fagerstrom CJ, Yvon AM, Wadsworth P. Cell cycle-dependent changes in microtubule dynamics in living cells expressing green fluorescent protein-α tubulin. Mol Biol Cell 2001;12:971-80.
- Mitchison TJ. Microtubule dynamics and kinetochore function in mitosis. Annu Rev Cell Biol 1988;4:527-49.
- Wang LG, Liu XM, Kreis W, Budman DR. The effect of antimicrotubule agents on signal transduction pathways of apoptosis: a review. Cancer Chemother Pharmacol 1999;44:355-61.
- Tong YG, Zhang XW, Geng MY, Yue JM, Xin XL, Tian F, et al. Pseudolarix acid B, a new tubulin-binding agent, inhibits angiogenesis by interacting with a novel binding site on tubulin. Mol Pharmacol 2006;69:1226-33.
- Wong VK, Chiu P, Chung SS, Chow LM, Zhao YZ, Yang BB, et al. Pseudolaric acid B, a novel microtubule-destabilizing agent that circumvents multidrug resistance phenotype and exhibits antitumor activity in vivo. Clin Cancer Res 2005;11:6002-11.