
Growth suppression and radiosensitivity increase by HMGB1 in breast cancer1
Introduction
HMGB1 (high-mobility group box-1, amphoterin, formerly named high-mobility group (HMG)1, belonging to the HMG protein family, is a cytokine-like, 215 amino acid nuclear protein, which is an important mediator of the body’s inflammation, ischemia, and injury[1,2]. HMGB1 is structured into 3 domains: 2 basic HMG boxes (HMG domains A and B) and a highly acidic C-terminal domain, which confer an overall dipolar appearance to this protein. Each of the HMG boxes is formed by 2 short and 1 long a-helix, that upon folding produce an L-or V-shaped 3-D domain structure. The concave surface of the L-or V-shaped HMG box domain contacts the DNA in the minor groove in 2 slightly different ways introducing important modifications in the structure of DNA, in particular a strong bend[1,2]. Presumably, these features are of relevance for the biological functions in which HMGB1 has been involved (DNA repair, recombination, replication, and transcription)[1,2]. HMGB1 can interact through its HMG box domains with a broad range of proteins ranging from nuclear cell proteins to viral proteins, including the recombination activation gene protein, several transcription factors, including the cellular tumor suppressor p53, octamer transcription factors Oct1, Oct2, Oct4, and Oct6, some homeotic HOX proteins, the steroid receptors, the general initiation factor human TATA-binding protein (hTBP), and the viral replication proteins Rep78 and Rep68[1,2]. HMG box A is important for binding to hTBP and p53, whereas the binding to Oct factors, HOX factors, and hormone receptors can take place through boxes A or B. Additionally, it has been found that HMGB1 was abundantly expressed in breast cancer tissues compared to normal breast tissues by a breast tissue microarray[3]. However, the critical roles of HMGB1 in breast cancer still need to be explored.
The retinoblastoma (Rb) gene, encoding a 928 amino acid phosphoprotein, is a tumor suppressor, and the loss or inactivation of Rb gene activity is seen as contributing to a broad range of tumors, including breast cancer[4,5]. RB plays important roles in the regulation of cell proliferation, cell cycle progression, apoptosis, telomerase activity, and so on[5?9]. At least 4 distinct protein-binding domains of RB have been identified and extensively characterized, including the A/B pocket, the large A/B pocket, the C-pocket, and the N-terminal domain[5,6]. The activated (hypo-phosphorylated) RB protein inhibits cell cycle progression from G1→S, in part via an interaction between the large A/B pocket of RB and the activation domains of the E2F family transcription factors, resulting in the repression of E2F target genes[7]. The cell cycle inhibitory activity of RB is regulated via interactions of the standard A/B-binding pocket domain of RB with the LXCXE ((where L=leucine, C=cysteine, E=glutamic acid and X=any amino acid) motif of target proteins. For example, interactions between RB and cell cycle regulatory proteins (G1/S cyclins and Cyclin-dependent kinases) and viral oncoproteins (Simian virus 40 large T antigen, adenovirus E1A, and human papillomavirus E7) inactivate the cell cycle inhibitory activity of RB and mediate its transcriptional repression[8]. Thus, the A/B pocket is likely to play an important role in RB tumor suppressor functions[5,6]. The A/B and C domains are conserved in 2 other Rb gene family proteins, p107 and p130, which also bind to LXCXE and regulate cell cycle-dependent transcription[10]. The activities of p107 and p130 overlap with, but are not identical to RB, and these proteins may partially substitute for RB functions. The standard A/B-binding pocket, which regulates the phosphorylation state and cell cycle regulatory activity of RB, is the site of most tumor-associated Rb mutations[4,5].
Sequence analysis suggests that the HMGB1 protein contains a consensus RB-binding LXCXE motif (amino acid 104?108). Therefore, in this study we have studied the potential association of HMGB1 and RB and the in vitro and in vivo activities of HMGB1 in human breast cancer cells, and demonstrated that the HMGB1 protein associated with the RB protein, the LXCXE motif was required for the interaction. We found that enhanced expression of HMGB1 suppressed in vitro cell growth and reduced in vivo tumor growth in a RB-dependent fashion. Finally, we found that enhanced expression of HMGB1increased sensitization of breast cancer cells to ionizing radiation, which was via a RB-independent mechanism.
Materials and methods
Cell culture and irradiation Human breast cancer cell lines MCF-7 (containing wild-type RB) and BT-549 (containing RB deletion mutation), and human osteoblastic cell line SAOS-2 (containing negative RB) were originally purchased from the American Type Culture Collection (Manassas, VA, USA) and maintained in Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA, USA) supplemented with 5% fetal bovine serum, 100 unit/mL penicillin, 100 µg/mL streptomycin, and a mixture of non-essential amino acids (Sigma-Aldrich, St Louis, MO, USA). The cell lines were incubated in a humidified atmosphere of 95% air and 5% CO2. RB+/+ and RB?/? MEF (mouse embryonic fibroblasts) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal calf serum (FCS), and all experiments were conducted using early passage (< 6 passages) MEF. Irradiation was performed with γ-radiation (JL Shepherd Mark I Radiator) using a 137Cs source emitting at a fixed dose rate of 3.5 Gy/min.
Transfection To establish stable transfections, the cells in 100 mm Petri tissue culture dishes at about 50%?60% confluence, were incubated overnight with 5 µg plasmid DNA, using Lipofectamine 2000 (Invitrogen, USA), according to the manufacturer’s instructions. The transfected cells were incubated in medium containing G418 (0.5 mg/mL), the G418-resistant colonies were observed approximately 3 weeks after transfection.
HMGB1 and RB vectors The wild-type HMGB1 expression plasmid (wtHMGB1) was created by cloning the full-length HMGB1 cDNA into a mammalian expression vector pCMV-Tag2B (Invitrogen, USA). LXCXE-defective HMGB1 (HMGB1-RXRXH, in which the LXCXE changed into the RXRXH) and LXCXE-truncated HMGB1 (HMGB1ΔLXCXE, in which the LXCXE was deleted) expression vectors were created by modification of the wtHMGB1 cDNA in the pCMV-Tag2B vector by a MORPH site-directed plasmid DNA mutagenesis kit (Stratagene, La Jolla, CA, USA). pGEX5X-RB was described in elsewhere[11]. The cyclin A reporter plasmid was obtained by cloning into the KpnI and HindIII restriction sites of the pGL2Basic vector with a 213 bp fragment of the human cyclin A promoter (from -165 to +48 bp, relative to the most 3' transcription initiation site), which was generated by PCR with oligonucleotides 5'-CTCCGGTACCAGCCAGTTTGTTTCTC-3' and 5'-TGGCAAGCTTAAGACGCCCAGAGATG-3'.
Generation of recombinant HMGB1 adenoviruses The E1-deleted adenovirus-β-galactosidase (Ad-β-gal) was obtained from Introgen Therapeutics (Houston, TX, USA). A recombinant adenovirus (pAd/CMV/V5-DEST, Invitrogen, USA) containing a DNA fragment encoding the complete amino acid sequence of human HMGB1 (Ad5-HMGB1), HMGB1-RXRXH (Ad5-HMGB1-RXRXH) or HMGB1ΔLXCXE (Ad5-HMGB1ΔLXCXE) between the CMV promoter (pCMV) and the polyadenylation signal (TK pA) was prepared. These adenoviral vectors were propagated in 293 human embryonic kidney cells (Invitrogen, USA) using the Stratagene MBS mammalian transfection kit (USA) with a modified calcium phosphate transfection protocol. The transfected cells were incubated at 37 °C for 7 d, then harvested and subjected to 4 freeze (liquid nitrogen)/thaw (a 37 °C water bath) cycles. The cell lysates were centrifuged at 12 000×g for 10 min at 4 °C, and the supernatant (primary virus stock) was transferred to a fresh screw-cap mini-centrifuge tube and stored at -80 °C. Recombinant adenoviruses were further amplified using the same procedure; the cell lysates were centrifuged on cesium chloride step gradients at 60 000×g at 4 °C for 2 h to separate viruses from defective particles and empty capsids. Recovered virus bands were dialyzed 3 times against phosphate-buffered saline (PBS). The viruses were aliquoted in a buffer containing 10 mmol/L Tris, pH 7.4, 10 mmol/L MgCl2, and 10% v/v glycerol) and stored at -80 °C. Under these conditions, there was no precipitation of virus particles or loss of virus infectivity due to inactivation or aggregation. To control the biological effect of the virus per second, the vector, Ad5.CMV.Null, expressing no transgene (Ad5), was constructed in a similar manner, but without subcloned gene sequences.
In vitro cell growth kinetics To assess in vitro cell proliferation, the infected cells were inoculated into 6-well dishes at 3×104 cells/well in 5.0 mL complete growth medium (DMEM plus 5% FCS) on d 0. For each clone tested, duplicate wells were counted by a hemocytometer on d 1-8. The duplicate cell counts were within ±5% SEM of the mean values.
MTT (3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide) assay of cell viability MTT assays are based on the ability of viable cells to convert MTT dye (3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide), a soluble tetrazolium salt (thioazyl blue) into an insoluble formazan precipitate, which is quantitated by spectrophotometry following solubilization in dimethyl sulfoxide[12]. Briefly, subconfluent proliferating cells in 96-well dishes were treated with a cytotoxic drug in standard growth medium, washed vigorously to remove the drug, and then postincu-bated for 48 h in fresh, drug-free culture medium. At this time, the cells were solubilized and absorbance readings were taken using a Dynatech 96-well spectrophotometer (Billingshurst, UK). The amount of MTT dye reduction was calculated based on the difference between absorbance at 570 and at 630 nm. Cell viability was expressed as the amount of dye reduction relative to that of the untreated control cells. Ten replicate wells were tested per assay condition, and each experiment was repeated at least 3 times.
Clonogenic assay The cells were trypsinized immediately after irradiation and counted. Known numbers were subcultured in 100 mm culture dishes in 2 sets of triplicates for each dose of radiation; sufficient numbers were seeded to ensure that about 50?100 macroscopic colonies would appear in each plate of untreated and uninfected control cells at the end of the 15 d. Colonies were then fixed, stained, and counted. Surviving fractions were normalized by the plating efficiency of unirradiated controls (30%-50% for MCF-7 and 40%-60% for BT-549).
Flow cytometry assay of cell cycle and apoptosis Cell cycle progression and apoptosis analysis was performed by flow cytometry. The culture medium was collected into centrifuge tubes. The cells removed by trypsin were poured into the same tubes. The cells were centrifuged for 5 min at 900×g. The supernatant were poured out, washed once with 1×PBS and centrifuged for another 5 min. The cells were finally fixed by 5 mL precooled 70% ethanol for at least 4 h. The fixed cells were centrifuged and washed with 1×PBS. After being centrifuged, the cell pellets were resuspended in 500 μL propidium iodine (10 μg/mL) containing 300 μg/mL RNase (Sigma-Aldrich, USA). Then the cells were incubated on ice for 30 min and filtered with a 53 μm nylon mesh. Cell cycle distribution was calculated from 10 000 cells with ModFit LT software (Becton Dickinson, San Joes, CA, USA) using FACSCaliber (Becton Dickinson, USA).
Immunoprecipitation Subconfluent proliferating cells in 150 cm2 dishes were harvested, and nuclear extracts were prepared, as described earlier[11,13]. Each immunoprecipitation (IP) was carried out using 6 μg antibody or antibody combination and 500 μg nuclear extract protein. The precipitated proteins were collected using protein G beads, washed, eluted in boiling Laemmli sample buffer, and subjected to Western blotting. The HMGB1 IP antibody was antibody T32, kindly provided by Dr Kimitoshi KOHNO, University of Occupational and Environmental Health, Fukuoka, Japan. The control IP antibody was a normal mouse immunoglobulin G (IgG, Santa Cruz Biotechnology, Santa Cruz, CA, USA).
Western blotting Western blotting assays were performed as described earlier[11,12]. Equal aliquots of protein extract (100 μg/lane) were electrophoresed on SDS-PAGE, transferred to nitrocellulose membranes (Millipore, Bedford, MA, USA), and blotted with primary antibodies: a mouse monoclonal anti-HMGB1 antibody (R&D Systems, Minneapolis, MN, USA); a rabbit polyclonal anti-RB antibody (C-15, Santa Cruz Biotechnology, USA) or a mixture of a rabbit polyclonal anti-PRAP antibody (P7607, Sigma, USA) plus a rabbit polyclonal anti-PRAP antibody (214/215, cleavage site specific antibody, Sigma, USA). The appropriate secondary antibodies were used at a dilution of 1:2000. Blotted proteins were visualized using the enhanced chemiluminescence system (Amersham Life Sciences, Arlington Heights, IL, USA), with colored markers (Bio-Rad Labora-tories, Hercules, CA, USA) as molecular size standards. As an internal control for the amount of protein loaded, the same filter was also immunoblotted with a polyclonal α-actin antibody (I-19, Santa Cruz Biotechnology, USA).
Glutathione-S-transferase capture assays Glutathione-S-transferase (GST) capture assays were performed as described in our previous studies[11,13]. 35S-methionine-labeled proteins were prepared by in vitro transcription and translation, using the T3 promoter of the pCMV-Tag2B vector. The GST fusion proteins were generated from cDNA cloned into the GST vector (pGEX), expressed in Escheria coli, and purified by affinity chromatography. In vitro translated labeled proteins were incubated with either GST alone (negative control) or GST fusion proteins for 4 h at 48 ℃, recovered using GSH gluathione agarose beads, eluted in boiling Laemmli sample buffer, and analyzed by SDS-PAGE autoradiography. To confirm their expression, the GST fusion proteins were visualized by Western blotting, using an anti-GST mouse monoclonal antibody (B-14, 1:5000 dilution, Santa Cruz Biotechnology, USA).
Assays of transcriptional activity Proliferating cells at 50%±70% confluency in 24-well dishes were incubated overnight with 0.25 µg of each vector (unless otherwise indicated) in serum-free DMEM containing Lipofectamine 2000 (Invitrogen, USA). The total transfected DNA was kept constant by the addition of the control vector. The cells were washed, incubated in serum-free, phenolphthalein-free DMEM (0.2 µL/well) for 24 h, and harvested for luciferase assays. To control transfection efficiency, plasmid pRSV-β-gal was cotransfected to allow normalization of luciferase values to β-gal activity in the same sample. Values were mean±SEM of 4 replicate wells and were representative of 3 independent experiments.
Measurement of caspase-3 activity The caspase-3 activity assay kits purchased from BioVison (Mountain View, CA, USA) were used for the detection of caspase-3 activity following the protocol recommended by the manufac-turer. In brief, proteolytic reactions were done containing cytosolic extracts, 2×reaction buffer containing DL-dithio-threitol (DTT), and caspase-3 colorimetric substrate (DEVD-p-nitroanilide). The reaction mixture was incubated at 37 °C for 1-2 h, and then the formation of p-nitroanilide was measured at 405 nm using an ELISA reader.
Tumor studies In the studies of MCF-7 cells, female nu/nu BALB/c mice were implanted subcutaneously with 17β-estradiol-sustained release pellets (60 d release pellets; 0.72 mg estradiol; Innovative Research of America, Sarasota, FL, USA). The mice were inoculated subcutaneously with approximately 5×106 infected MCF-7 or BT-549 cells suspended in 0.3 mL Matrigel-DMEM. Inoculum size was selected to produce <100% tumor incidence under optimal conditions, based on our experience with these cells, so that we could detect either an increase or decrease in tumor incidence. The in vivo studies were independently performed twice. Tumor volumes were monitored weekly by caliper measurement of the length, width, and height, and were calculated using the formula for a semiellipsoid (4/3πr3/2) up to 3 weeks. Each group contained 10 mice; the data from 2 independent experiments were combined in the analysis.
Statistical analysis Cell viability and luciferase activity data were analyzed using Student’s paired t-test. Animal data were subjected to ANOVA with Statistica software (StatSoft, Berkeley USA) to identify individual differences. Comparisons were considered to be statistically significant when P<0.05.
Results
HMGB1 interacts with RB The sequence analysis shows that the central region of the HMGB1 protein contains a consensus RB-binding motif, LXCXE (aa104LFCSE108), thus we first assessed if the HMGB1 protein interacts with the RB protein by IP, followed by Western blotting. As shown in Figure 1A, the RB protein was easily detected in the HMGB1 IP of MCF-7 cells containing the wild-type Rb gene; but no RB protein was found in the HMGB1 IP of BT-549 cells that have Rb gene deletion. The HMGB1 protein was also detected in the RB IP in MCF-7 cells. The RB or HMGB1 protein was not found in the normal mouse IgG IP (as a negative control). These data indicate that endogenous HMGB1 and RB physically associate with each other in MCF-7 cells.
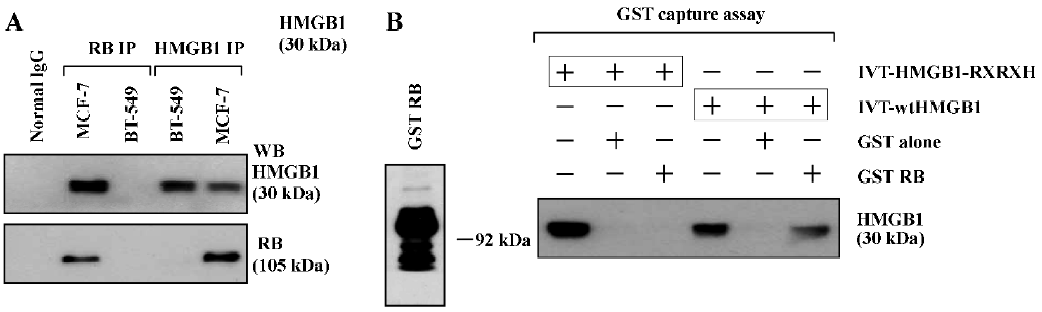
The direct interaction of HMGB1 and RB was also observed by GST capture assays. As shown in Figure 1B, the in vitro-translated (IVT) wtHMGB1 protein bound to beads coated with the GST-RB protein, but not to beads coated with the GST protein alone. However, IVT-HMGB1-RXRXH, in which 104LXCXE108 was mutated into 104RXRXH108 using a site-directed mutagenesis kit (Stratagene, USA), failed to bind to the GST-RB protein. These results suggest that the LXCXE motif of the HMGB1 protein is critical and essential for HMGB1 binding to RB.
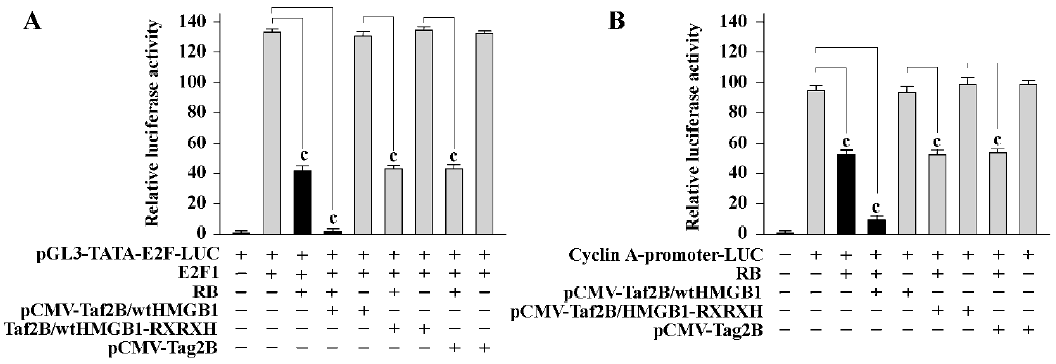
HMGB1 enhances RB-mediated transcription repression The ability to block cell cycle progression is intimately linked to the ability of RB to repress E2F-regulated transcription[13]. We next examined if HMGB1 showed any effects on RB-meditated E2F transcription repression. As shown in Figure 2A, the activation of an E2F reporter by E2F1 in RB-negative SAOS-2 cells was repressed by RB (69% reduction, P<0.01). The ability of RB in the transcription repression was enhanced by cotransfection of wtHMGB1 (about 97% reduction, P<0.005), but not affected by HMGB1-RXRXH or “empty”control pCMV-Tag2B vector alone; wtHMGB1 by itself did not result in any effects on E2F transcription activity. Similar results were obtained with HMGB1-mediated RB repression of cyclin A (Figure 2B). These studies suggest that HMGB1 can act as a cofactor for RB-mediated transcription repression, at least for E2F and cyclin A.
HMGB1 causes RB-dependent growth inhibition To further study the roles of HMGB1 in breast cancer cells, we tried to stably overexpress HMGB1 in 2 breast cancer cell lines, MCF-7 (containing wild-type RB) and BT-549 (containing mutant-RB) by transfection with pCMV-Tag2B/wtHMGB1) or pCMV-Tag2B/HMGB1-RXRXH. The transfected cells were postcultured in medium containing 500 µg/mL neomycin (G418), and the G418-resistant clones were viewed about 21 d following transfection. The formation of G418-resistant clones could be seen in the BT-549 cell line transfected with wtHMGB1, but no significant clones were viewed in plates with MCF-7 cells transfected with wtHMGB1. These findings suggest that the ectopic expression of HMGB1 inhibits the proliferation of functional RB-containing breast cancer cells, not mutant RB-containing cells. However, G418-resistant clones, formed in the culture plates of both MCF-7 and BT-549 cell lines and transfected with pCMV-Tag2B-HMGB1-RXRXH, indicating that the suppression of cell growth by HMGB1 requires HMGB1-RB interaction. In other words, the LXCXE motif is necessary for HMGB1 anticell growth.
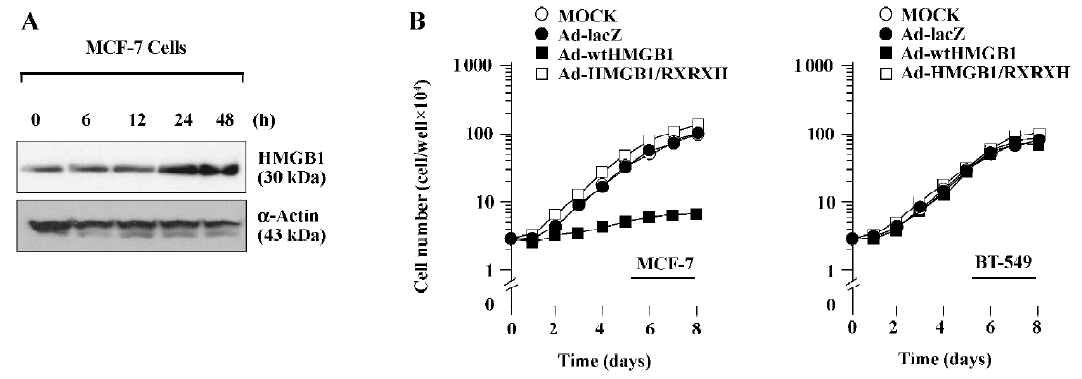
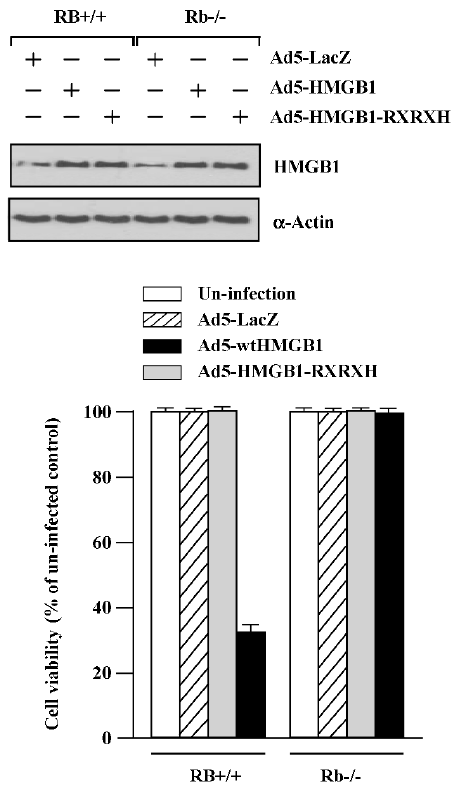
To further identify the antigrowth properties of HMGB1-expressing cells, we next produced an adenovirus expression system to achieve a high expression of HMGB1 levels. The cDNA encoding wtHMGB1 or HMGB1-RXRXH under the CMV promoter were inserted into the E1 region of E1- and E3-deleted adenovirus type 5 DNA encoding bacterial β-gal, Ad5CMV-lacZ. The replication-defective recombinant viruses, designated Ad5-wtHMGB1 or Ad5-HMGB1-RXRXH, were isolated by plaque purification, and high-titer virus stocks were produced. Preparations with a higher ratio exhibited increased cytotoxicity to breast cancer cells using a MOI of >100 pfu/cell (data not shown). As shown in Figure 3A, the time course of HMGB1 expression was determined in MCF-7 cells by immunoblotting after infection with Ad5-wtHMGB1 at a MOI of 100 pfu/cell. An increased expression of the HMGB1 protein was readily detected after 24 h infection. Similar results were observed by infection with Ad5-HMGB1-RXRXH and obtained with the employment of BT-549 cells (data not shown). The number of infected cells was then counted every day up to 8 d. The proliferation of the MCF-7 cells was inhibited by Ad5-wtHMGB1, starting at 72 h following infection, whereas the proliferation of BT-549 cells was not affected by Ad5-wtHMGB1 (Figure 3B). In contrast, Ad5-HMGB1-RXRXH caused no significant effects of MCF-7 cell growth. Similar results were also obtained in another 2 human breast cancer cell lines, T-47D containing wild-type RB (data not shown). Neither Ad5-wtHMGB1 nor Ad5-HMGB1-RXRXH influenced BT-549 cell proliferation. The similar studies were performed with mouse embryonic fibroblasts (MEF). As illustrated in Figure 4, Ad5-wtHMGB1 inhibited cell growth in Rb+/+ MEF, not in Rb?/? MEF derived from embryos isolated from the mating of mice with Rb+/?. Again, neither RB+/+ nor Rb?/? MEF responded to Ad5-HMGB1-RXRXH. Thus, these results indicate that the adenovirus-mediated transfer of the HMGB1 gene causes cell growth suppression in a RB-dependent manner.
HMGB1 causes RB-dependent G1 arrest and apoptosis induction To examine the mechanism(s) by which HMGB1 mediates growth suppression, we studied the impacts of HMGB1 on cell cycle progression and apoptosis induction. Increasing G1 cell cycle arrest and sub-G1 population (indicat-ing apoptosis) were observed in the MCF-7 cells infected with Ad-wtHMGB1 at a MOI of 100 pfu/cell for 72 and 96 h, as determined by flow cytometry assays (Figure 5). The sub-G1 population was accumulated with the increased time of Ad5-wtHMGB1 infection in the MCF-7 cells, that is, 5.4% and 13.5% for 72 and 96 h infection, respectively. In contrast, infection with Ad5-HMGB1-RXRXH or Ad5-lacZ caused no significant changes in the cell cycle profile of MCF-7 cells. No G1 arrest and sub-G1 population accumulation was induced in BT-549 cells infected with Ad5-wtHMGB1, Ad5-HMGB1-RXRXH or Ad5-lacZ (data not shown). Therefore, these findings indicate that RB is a determinant in the HMGB1-mediated G1 arrest and apoptosis, which may be important mechanisms contributing to the HMGB1 inhibition of cell proliferation.
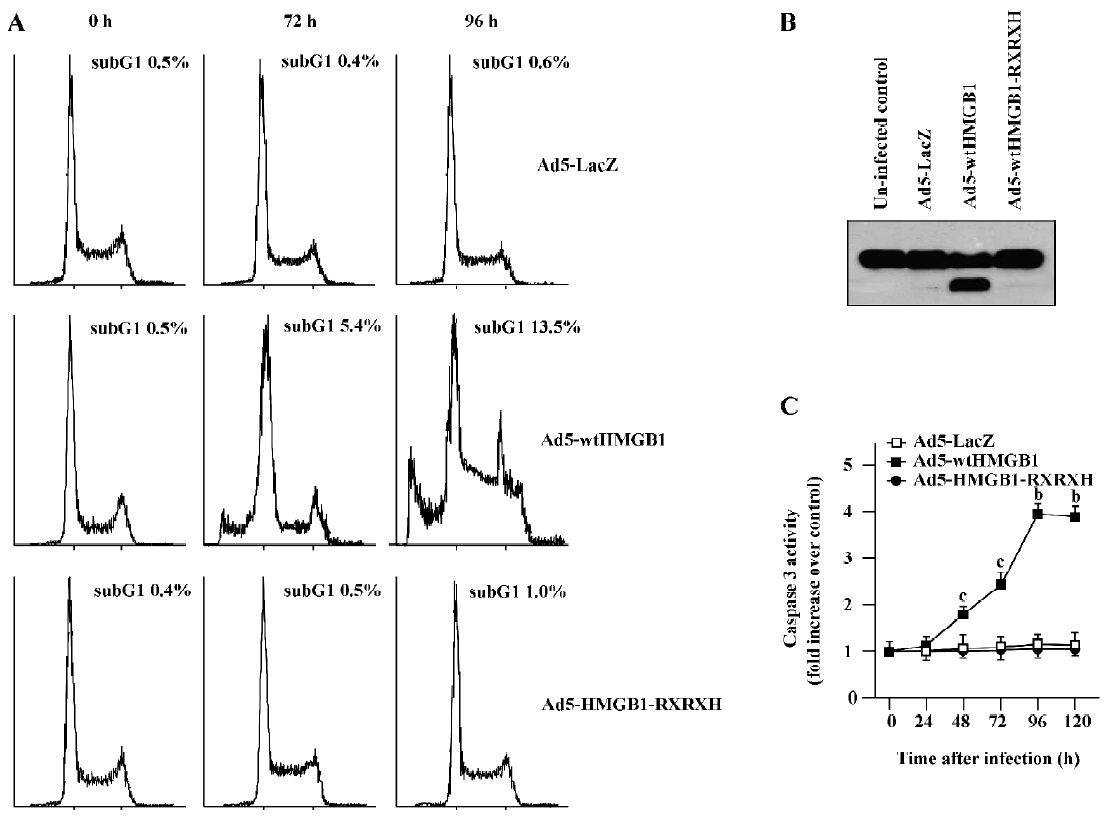
To examine the molecule mechanisms which may contribute to HMGB1-induced apoptosis, we determined if HMGB1 exhibited any affect on poly (ADP-ribose) polymerase (PARP) cleavage and caspase 3 activity. It has known that the proteolytic cleavage of PARP is 1 characteristic event of apoptosis. PARP is a nuclear enzyme involved in DNA repair, DNA stability, and transcriptional regulation. Caspases, in particular caspases-3 and -7, cleave the 116 kDa form of PARP-1 at the DEVD site to generate an 85 and 24 kDa fragment[14,15]. The cleavage of PARP-1 between Asp214 and Gly215 results in the separation of the 2 zinc-finger, DNA-binding motifs from the automodification and catalytic domains. Thus, PARP-1 cleavage has been considered a hallmark of apoptosis. As shown in Figure 5B, Ad5-HMGB1 caused a clear cleavage of PARP, while neither Ad5-HMGB1-RXRXH nor Ad5-LacZ affected PARP in the MCF-7 cells. Moreover, Ad5-HMGB1 increased caspase-3 activity in a time-dependent manner (Figure 5C). Therefore, although further studies are needed, these results indicate that the HMGB1↑→caspase-3↑→ PARP↑→apoptosis may be an important pathway for HMGB1-mediated apoptosis induction.
HMGB1 increases cell sensitivity to ionizing radia-tion To determine the effects of HMGB1 on the radiosensitivity of breast cancer cells, a MTT survival analysis was performed with the 2 human breast cancer cell lines MCF-7 and BT-549. Exponentially growing cells cultured in 6-well tissue culture dishes were uninfected or infected with Ad5-wtHMGB1, Ad5-HMGB1-RXRXH or Ad5-lacZ (a control) at a MOI of 100 pfu/cell only for 24 h, and then irradiated with various doses of γ-rays. The cells were subjected to MTT assay for cell survival 24 h following irradiation. As shown in Figure 6A, the sensitivity to γ-ray irradiation in the MCF-7 cells infected with Ad5-wtHMGB1 or Ad5-HMGB1–RXRXH was significantly higher than the uninfected cells or the cells infected with Ad5-lacZ. For example, 4 Gy of γ-ray irradiation reduced the cell viability to about 38%-40% in both the uninfected and Ad5-LacZ-infected control cells, while in the cells infected with Ad5-wtHMGB1 or Ad5-HMGB1-RXRXH, the cell viability was reduced to approximately 13%?15% (>3-fold, P<0.05, Student’s t-test). Twenty four hours of infection with Ad5-wtHMGB1, Ad5-HMGB1-RXRXH, or Ad5-lacZ at a MOI of 100 pfu/cell did not cause any loss of cell viability, consistent with no significant inhibition of MCF-7 cell growth by <48 h infection of Ad5-wtHMGB1 (Figure 3), therefore, the HMGB1 radiosensitivity was not just due to the addition of HMGB1 and irradiation cytotoxicity. Similar results were also observed with BT-549 cells infected with Ad5– wtHMGB1, although the BT-549 cells were slightly more resistant to γ-ray irradiation compared to MCF-7 cells.
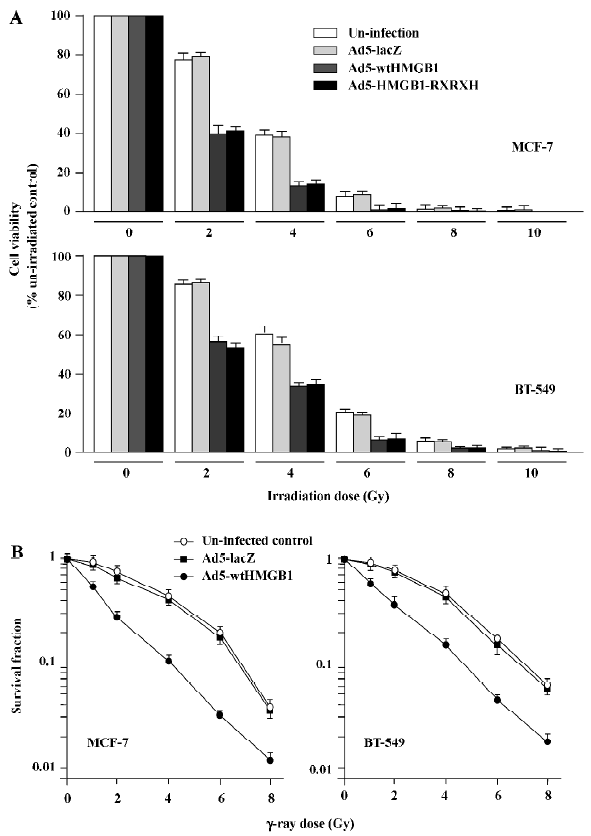
We also employed clonogenic assay to confirm radiosensitivity. Similar results with HMGB1-mediated radiosensitivity were observed, as shown in Figure 6B. The sensitivity enhancement ratio was 3.2 (ID50) and 1.6 (ID90) for MCF-7, and 2.6 (ID50) and 1.4 (ID90) for BT-549, respec-tively. These results indicate that the enhanced expression of HMGB1 results in a significant increase in radiosensitivity of human breast cancer cells. Moreover, such HMGB1-mediated radiosensitivity is independent on RB gene status, since radiosensitization by HMGB1 was observed in both the MCF-7 and BT-549 cell lines. The further investigation of the underlying mechanism(s) of HMGB1 radiosensitivity is ongoing in our laboratory.
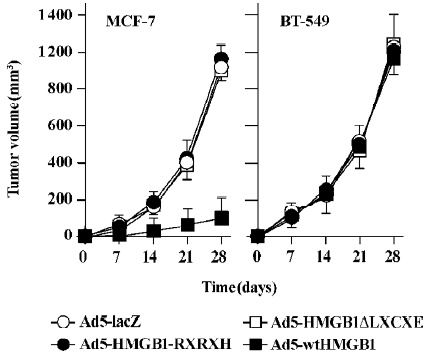
HMGB1 suppresses in vivo tumor growth To determine the in vivo antitumor activities of HMGB1, we investigated the tumorigenic potential of Ad5-HMGB1-infected MCF-7 and BT-549 cells compared with infections of Ad5-HMGB1-RXRXH, and Ad5-HMGB1ΔLXCXE was deleted using the MORPH site-directed plasmid DNA mutagenesis kit (Invitrogen, USA). As illustrated in Figure 7, tumor growth and size were markedly suppressed in mice inoculated with Ad5-wtHMGB1-infected MCF-7 cells (P<0.001). In contrast, Ad5-wtHMGB1-infected BT-549 tumor growth and size showed no significant difference from those in the tumors formed with uninfected cells or Ad5-lacZ-infected control cells (P>0.05). Ad5-HMGB1-RXRXH or Ad5-HMGB1ΔLXCXE tumors versus uninfected or Ad5-LacZ-infected control tumors were not statistically significant in both the MCF-7 and BT-549 tumor models. These data, consistent with the in vitro antitumor activity described earlier, suggest that HMGB1 may be a breast cancer growth suppressor through a mechanism that depends on the Rb gene status and HMGB1-RB interaction, that is, RB is an important determinant of HMGB1-mediated tumor suppression. The LXCXE motif of HMGB1 is essential for HMGB1 antitumor growth.
Discussion
In a series of experiments, we studied the in vitro and in vivo activities of HMGB1, a nuclear protein containing a consensus RB-binding LXCXE motif in breast cancer cell lines with different Rb gene status. HMGB1 as a pro-inflammatory cytokine has been implicated in the pathogenesis of a broad range of immune-mediated diseases, including arthritis[1,2]. The importance of this study demonstrated for the first time that HMGB1 can function as a tumor suppressor in breast cancer cells via a RB-dependent pathway. First, we discovered a physical interaction between the HMGB1 and RB protein; this association is via a strong LXCXE-dependent mechanism (Figure 1). It is known that a number of endogenous proteins that interact with RB also contain a LXCXE or LXCXE-like sequence, such as histone deacetylase (HDAC)-1 and HDAC2, and ATPase and BRM/SWI2-related gene (BRG1) from the SWI/SNF nucleosome remodeling complex. The oncoproteins of several DNA tumor viruses, including E1A from adenovirus E7 from human papillomavirus and T antigen from SV40, inactivate the ability of RB to suppress cell growth via directly binding to the LXCXE binding site in the RB protein. Moreover, the mutation of the LXCXE sequence in these proteins prevents their inhibitory effect on Rb and their ability to transform cells, indicating the importance of the LXCXE binding site. Therefore, further studies in our laboratory are ongoing to determine if HMGB1 blocks or reduces the binding capability of these DNA tumor virus proteins to RB by competing for the LXCXE binding site.
Second, we observed that the increased expression of HMGB1 enhanced the RB-mediated suppression of E2F and cyclin A transcriptional activation (Figure 2); however, HMGB1 by itself does not affect E2F and cyclin A transcriptional activity. It is known that RB represses transcription by at least 2 different mechanisms: it can bind transcription factors, such as E2F, and cyclin A can block their ability to activate transcription; and the Rb-E2F repressor complex that forms at promoters can actively repress transcription. This cooperativity of HMGB1 and RB is likely to be the result of the direct interaction between HMGB1 and RB for 2 reasons: the RB-binding LXCXE motif of the HMGB1 sequence is required for the cooperation, and RB is able to contact both HMGB1 and E2F1 simultaneously. It will be interesting to know if HMGB1 is complexed with E2F1 or cyclin A.
Third, the high HMGB1 expression caused G1 arrest and sub-G1 (apoptotic cell) accumulation in MCF-7 cells, not BT-495 cells (Figure 5), suggesting that the anticell proliferation of HMGB1 may have been a result of G1 cell cycle arrest and apoptosis induction. HMGB1-mediated apop-tosis may be due to caspase-3 induction and subsequent PARP cleavage. It is known that the repression of E2F-containing promoters by RB is considered to be one of the key mechanisms by which RB induces G1 arrest. The ability of HMGB1 to cooperate with RB in the repression of E2F1 may therefore be an underlying mechanism for the observed cooperation between RB and HMGB1 in the induction of G1 arrest. The LXCXE motif, RB-binding region of HMGB1 is also important for HMGB1 G1 arrest functions and apoptosis.
Fourth, we found that an increase of HMGB1 expression by either transfection of the pCMV-Tag2/wtHMGB1 mammalian expression vector or the adenovirus-mediated HMGB1 gene (Ad5-wtHMGB1) expression significantly inhibits cell proliferation of MCF-7 containing wild-type RB, rather than BT-495 cells containing deletion mutant RB (Figures 3,4). Ad5-wtHMGB1 significantly delays tumor growth in nud mice (Figure 7). All of these activities of HMGB1 required the interaction of HMGB1 and RB, since HMGB1-RXRXH (inactivating LXCXE mutant) or HMGB1ΔLXCXE (LXCXE deletion mutant) failed to exhibit these antitumor activities. In other words, the consensus RB-binding LXCXE motif is critical and essential for the antitumor activities of HMGB1. Therefore, a potential mechanism for HMGB1 antitumor activity is proposed: HMGB1↑→HMGB1-RB interaction→E2F or cyclin D1 transcription activity↑→ G1 arrest and apoptosis induction of cell growth inhibition.
Finally, we demonstrated that the elevated expression of HMGB1 by either Ad5-wtHMGB1 or Ad5-HMGB1-RXRXH significantly increased cellular sensitivity to ionizing radiation in both wild-type RB MCF-7 cells and RB mutant BT-549 cells (Figure 6), indicating that HMGB1-mediated radiosensitivity does not require the interaction of HMGB1 and RB, that is, via a RB-independent mechanism. These exciting results suggest that HMGB1, a well-known pro-inflammatory cytokine, also functions as a tumor suppressor and a radiosensitizer in human breast cancer, supporting additional studies of the potential of the in vivo application of HMGB1 gene therapy in breast cancer patients. Further studies in our laboratory are underway to determine if HMGB1 affects DNA strand break and repair as a result of radiation, and the expression of DNA damage repair genes.
Breast cancer is the second leading cause of death from cancer in women worldwide, with more than 240 000 new patients every year. RB has been reported to be aberrant in approximately 20%-35% of breast cancers[16,17] and has been associated with poor disease outcome. Furthermore, loss of heterozygosity or other alterations at the Rb locus are often observed in primary breast cancer specimens[18?20]. The RB activities are regulated (activated or inactivated) by a large number of RB-interacting proteins. For example, the overproduction of cyclin D1 and cyclin E, which mediate the inactivation of RB, are very common events in breast cancer[21], and the deregulation of E2F target genes can be associated with poor prognosis in certain breast cancer cases[21]. These findings suggest that RB is an important tumor suppressor in breast cancer. Therefore, based on the observations in this study, further exploration of HMGB1 functions as a novel RB-associated protein will be significant in understanding RB-related pathogenesis of tumor progression and for identifying a new target for biological therapy in breast cancer.
References
- Yang H, Wang H, Czura CJ, Tracey KJ. HMGB1 as a cytokine and therapeutic target. J Endotoxin Res 2002;8:469-72.
- Wang H, Tracey KJ. High mobility group box 1 (HMGB1). Crit Care Med 2005;33:S472-4.
- Brezniceanu ML, Volp K, Bosser S, Solbach C, Lichter P, Joos S, et al. HMGB1 inhibits cell death in yeast and mammalian cells and is abundantly expressed in human breast carcinoma. FASEB J 2003;17:1295-7.
- Classon M, Harlow E. The retinoblastoma tumour suppressor in development and cancer. Nat Rev Cancer 2002;2:910-7.
- van Deursen JM. Rb loss causes cancer by driving mitosis mad. Cancer Cell 2007;11:1-3.
- Delston RB, Harbour JW. Rb at the interface between cell cycle and apoptotic decisions. Curr Mol Med 2006;6:713-8.
- Stevaux O, Dyson NJ. A revised picture of the E2F transcriptional network and RB function. Curr Opin Cell Biol 2002;14:684-91.
- Helt AM, Galloway DA. Mechanisms by which DNA tumor virus oncoproteins target the Rb family of pocket proteins. Carcinogenesis 2003;24:159-69.
- Gabellini C, Del Bufalo D, Zupi G. Involvement of RB gene family in tumor angiogenesis. Oncogene 2006;25:5326-32.
- De Falco G, Giordano A. pRb2/p130: a new candidate for retinoblastoma tumor formation. Oncogene 2006;25:5333-40.
- Fan S, Yuan R, Ma Y, Xiong J, Meng Q, Erdos MR, et al. Disruption of the BRCA1 LXCXE motif alters BRCA1 function activity and regulation of RB family but not RB protein binding. Oncogene 2001;20:4827-41.
- Fan S, Gao M, Meng Q, Laterra JJ, Symons MH, Coniglio S, et al. Role of NF-kappaB signaling in hepatocyte growth factor/scatter factor-mediated cell protection. Oncogene 2005;24:1749-66.
- Fan S, Ma Y, Wang C, Yuan R, Xiong J, Meng Q, et al. Role of direct interaction in BRCA1 inhibition of estrogen receptor activity. Oncogene 2001;20:77-87.
- Koh DW, Dawson TM, Dawson VL. Mediation of cell death by poly (ADP-ribose) polymerase-1. Pharmacol Res 2005;52:5-14.
- Rosen A, Casciola-Rosen L. Macromolecular substrates for the ICE-like proteases during apoptosis. J Cell Biochem 1997;64:50-4.
- Pietilainen T, Lipponen P, Aaltomaa S, Eskelinen M, Kosma VM. Syrj鋘en K. Expression of retinoblastoma gene protein (Rb) in breast cancer as related to established prognostic factors and survival. Eur J Cancer 1995;31A:329-33.
- Oesterreich S, Fuqua SA. Tumor suppressor genes in breast cancer. Endocr Relat Cancer 1999;6:405-19.
- Chano T, Kontani K, Teramoto K, Okabe H, Ikegawa S. Truncating mutations of RB1CC1 in human breast cancer. Nat Genet 2002;31:285-8.
- Borg A, Zhang QX, Alm P, Olsson H, Sellberg G. The retinoblastoma gene in breast cancer: allele loss is not correlated with loss of gene protein expression. Cancer Res 1992;2:2991-4.
- Ceccarelli C, Santini D, Chieco P, Taffurelli M, Gamberini M, Pileri SA, et al. Retinoblastoma (RB1) gene product expression in breast carcinoma. Correlation with Ki-67 growth fraction and biopathological profile. J Clin Pathol 1998;51:818-24.
- Malumbres M, Barbacid M. To cycle or not to cycle: a critical decision in cancer. Nat Rev Cancer 2001;1:222-31.
- Dai H. A cell proliferation signature is a marker of extremely poor outcome in a subpopulation of breast cancer patients. Cancer Res 2005;65:4059-66.