
Fluvastatin inhibits activation of JAK and STAT proteins in diabetic rat glomeruli and mesangial cells under high glucose conditions1
Introduction
Diabetic nephropathy is the main cause of end-stage renal disease requiring dialysis. A basic mechanism underlying diabetic nephropathy appears to be the high glucose (HG)-induced overexpression of transforming growth factor-β (TGF-β) and the accumulation of extracellular matrix (ECM) molecules, such as collagen IV and fibronectin[1,2]. For example, glomerular mesangial cells (GMC) showed increased levels of TGF-β and ECM when cultured in the presence of HG[1,2]. Recent studies suggest that Janus kinase (JAK)/signal transducers and activators of transcription (STAT) signaling cascades may contribute to diabetic nephropathy[3]. This pathway is mainly related to renal cell growth, production of the cytokine TGF-β, as well as the ECM proteins collagen IV and fibronectin[4].
JAK/STAT is an important signaling pathway, which is known to mediate the signaling of numerous cytokines and growth factors and is implicated in the regulation of a wide range of cellular processes, such as proliferation, differentiation, and apoptosis[5]. The JAK enzymes, JAK1, JAK2, JAK3, and tyrosine kinase-2 (TYK2), are responsible for the activation of the STAT (STAT1, STAT2, STAT3, STAT4, STAT5A/B, and STAT6), which are latent cytoplasmic transcription factors. STAT, when activated by tyrosine and/or serine phosphorylation, form homo- and heterodimers and translocate to the nucleus where they regulate the expression of various genes involved in cellular proliferation[6].
It was reported that activation of JAK/STAT was induced by HG and angiotensin II (Ang II) in rat GMC cultured in vitro[7]. That is, the exposure of GMC to hyperglycemia induces the tyrosine phosphorylation of JAK2, which was accompanied by the tyrosine and/or serine phosphorylation of STAT1, STAT3, and STAT5[7]. In addition, studies have also shown that the activation of JAK2 was essential for both Ang II- and hyperglycemia-induced collagen IV production and GMC growth[7]. Wang et al[4] reported that the activation of JAK2 and STAT1 proteins was a requirement for the hyperglycemia-induced production of TGF-β and fibronectin in GMC. The exposure of vascular smooth muscle cells (VSMC) to HG induced tyrosine phosphorylation of JAK2, which was accompanied by the tyrosine and/or serine phosphorylation of STAT1 and STAT3 and increased VSMC proliferation[8]. In addition, the activation of JAK/STAT also detected in diabetic rat glomeruli, and treatment with JAK2 inhibitor AG490 lowered systolic blood pressure and significantly reduced urinary protein excretion[9]. Therefore, it appears that the activation of JAK2 and STAT proteins by hyperglycemia might play an important role in both promoting cell proliferation and the synthesis of ECM molecules. The activation of JAK/STAT may be one of the major mechanisms involved in high glucose-induced glomerular injury[4].
Accumulating evidence has suggested that 3-hydroxy-3-methylglutanyl coenzyme A (HMG-CoA) reductase inhibitors (statins) have anti-inflammatory and endothelial cell-protective actions that are independent of the cholesterol-lowering effect[10]. Statins may influence intracellular pathways that are involved in the inflammatory and fibrogenic responses in progressive renal injury[10]. Fluvastatin is effective in improved renal function in streptozotocin (STZ)-induced diabetic rats[11]. Fluvastatin significantly suppressed proteinuria and serum creatinine in rats with puromycin aminonucleoside (PAN) nephrosis[12]. Fluvastatin could inhibit the Ang II-induced activation of JAK/STAT in VSMC[13]. A recent study demonstrated that simvastatin blocked the HG- and Ang II-induced activation of the JAK/STAT pathway in GMC[14]. Does fluvastatin show a similar physiological role? In the present study, we used both in vivo and in vitro methods to examine the effects of fluvastatin on the HG-induced activation of JAK2 and STAT proteins.
Materials and methods
Reagents Fluvastatin sodium was provided by Beijing Novartis (Beijing, China). D-glucose, D-mannitol and STZ were obtained from Sigma (St Louis, MO, USA). AG490 was purchased from Calbiochem-Novabiochem (La Jolla, CA, USA). The antibodies against SHP-1, JAK2, STAT1, STAT3, phospho-STAT1 (A-2), phospho-STAT3 (B-7), phospho-Tyr (PY99), protein A/G-agarose, and the enhanced chemiluminescence detection kit were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). SHP-2 and phospho-SHP-2 (Tyr580) were purchased from Cell Signaling Technology (Beverly, MA, USA). All culture media was purchased from Gibco-BRL (Grand Island, NY, USA). TRIzol reagent was obtained from Invitrogen Life Technologies (Carlsbad, CA, USA). The RT-PCR system was obtained from Promega (Madison, WI, USA). TGF-β1 and the fibronectin ELISA systems were purchased from R&D Systems (Minneapolis, MN, USA), and the polyvinylidene difluoride (PVDF) membrane was purchased from Millipore (Billerica, MA, USA).
STZ-induced diabetes Five-week-old male Sprague–Dawley rats (Grade II, Certificate N
Isolation of glomeruli The renal cortex was minced with a razor blade and pressed against a 0.3 mm stainless steel grid. Large fibrous tissues were retained on the grid surface, whereas glomeruli and tubular segments passed through. The glomeruli were then isolated by filtration through a 75 µm nylon mesh using an ice-cold 0.9% NaCl solution[15]. Those retained on the sieve were collected, washed by centrifugation (4 °C, 2000×g), and suspended in 50 mmol/L Tris-HCl (pH 7.4). The tissues were maintained at 4 °C during the entire isolation procedure. The purity of the glomerular suspensions was then assessed by light microscopy and estimated to be at least 95% glomeruli at the end of each preparation. The glomerular suspensions were then centrifuged at 4000×g for 5 min and used to extract total protein and RNA.
Histology The kidneys were fixed in 4% formaldehyde and embedded in paraffin; 4 µm sections were prepared and stained with hematoxylin–eosin and periodic acid-Schiff. The renal cortex sections were observed under light microscopy using the computer image analysis system. At least 50 glomeruli per animal were examined. Glomerular diameter was measured using TD2000 image analysis software (Tiandi-bainian, Beijing, China).
Cell culture The GMC were obtained from intact glomeruli of 5-week-old Sprague-Dawley rats and characterized according to published methods[16]. The GMC were plated on plastic tissue culture flasks in RPMI-1640 medium with 20% fetal bovine serum, 100 U/mL penicillin, 100 µg/mL streptomycin, and 0.6 U/mL insulin. GMC of passages 5–10 were grown to 75%–85% confluence, washed once with serum-free RPMI-1640 medium, and then growth-arrested in serum-free RPMI-1640 medium for 24 h to synchronize the cell growth. The GMC were divided into 5 groups: (i) normal glucose (NG, 5.5 mmol/L); (ii) NG+mannitol (24.5 mmol/L); (iii) HG (30 mmol/L); (iv) HG+AG490 (10 µmol/L); and (v) HG+fluvastatin (1 µmol/L).
Protein extraction Isolated glomeruli or GMC were washed twice with ice-cold PBS with 1 mmol/L Na3VO4, and then treated for 60 min with ice-cold lysis buffer [20 mmol/L Tris-HCl, pH 7.4, 2.5 mmol/L EDTA, 1% Triton X-100, 10% glycerol, 1% deoxycholate, 0.1% SDS, 10 mmol/L Na4P2O7, 50 mmol/L NaF, 1 mmol/L Na3VO4 and 1 mmol/L phenyl-methanesulfonyl fluoride (PMSF)]. The cell lysates were centrifuged at 14 000×g for 25 min at 4 °C and the supernatants were collected. The protein concentration was measured by Bradford’s method.
Immunoprecipitation and Western blotting study of JAK2 and SHP-1 The cell lysates were incubated with 10 µg/mL of either anti-JAK2 or anti-SHP-1 antibody at 4 °C for 2 h and precipitated by an addition of 20 µL protein A/G agarose beads at 4 °C overnight. The immunoprecipitates were washed 3 times with ice-cold wash buffer (10 mmol/L Tris-HCl, pH 7.4, 150 mmol/L NaCl, 0.1% Triton X-100, 1 mmol/L PMSF, and 1 mmol/L Na3VO4). The immunoprecipitated proteins were subjected to SDS-PAGE and then transferred to a PVDF membrane. After blocking in 5% non-fat milk for 2 h at 37 °C, the membrane was incubated with anti-phosphotyrosine antibody (1:1000) overnight at 4 °C, followed by incubation with the secondary antibodies conjugated with horseradish peroxidase (1:10000) for 2 h at room temperature. After extensive washing, the membrane was exposed to X-ray film using an enhanced chemiluminescence detection kit. The intensity of the bands was measured using LabWorks 4.5 (UVP, Upland, CA, USA).
Western blotting studies of SHP-2 and STAT proteins The cell samples were resolved by SDS–PAGE, transferred to a PVDF membrane, and blocked with 5% non-fat milk for 2 h at 37 °C. The membrane was incubated overnight at 4 °C with anti-SHP-2 (1:500) and anti-STAT (1:400) antibodies, respectively. Subsequently, the membranes were washed 3 times for 10 min each with TBS-T (10 mmol/L Tris-HCl, 150 mmol/L NaCl, 0.05% Tween 20) and incubated for 90 min with goat anti-rabbit IgG or goat anti-mouse IgG horseradish peroxidase conjugate. After extensive washing, the membranes were exposed to X-ray film and results were analyzed as described previously.
TGF-β1 RT-PCR analysis Total RNA and then cDNA were prepared from rat glomeruli and cultured cells using TRIzol reagent and RT-PCR kits. PCR was carried out for 30 cycles according to the following procedure: 95 °C for 60 s, 57 °C for 50 s, and 72 °C for 60 s; The primers for TGF-β1 and GAPDH were 5'-CCA TGA CAT GAA CCG ACC CT-3', 5'-CCG GGT TGT GTT GGT TGT AG -3' and 5'-TAT CGG ACG CCT GGT TAC-3', 5'-CTG TGC CGT TGA ACT TGC-3', respectively. The PCR products were subjected to 2% agarose gel electrophoresis and analyzed with a GDS-8000 Bioimaging system (UVP, upland, CA, USA) and LabWorks 4.5 software. RNA expression was quantified by comparison with internal-control GAPDH.
TGF-β1 and fibronectin ELISA After the cells were cultured in 6-well plates with or without different stimuli for 48 h, the supernatants were collected and TGF-β1 and fibronectin levels were measured by an ELISA kit, according to the manufacturer’s instruction. The TGF-β1 protein was quantified using the commercial Quantikine TGF-β1 ELISA kit. Briefly, the cell supernatants were activated with 1 mol/L HCl for 10 min, followed by neutralization with 1.2 mol/L NaOH. The activated samples were applied to the plate precoated with soluble type II receptor and incubated at room temperature for 3 h. After extensive washing, horseradish peroxidase-conjugated anti-TGF-β1 antibody was added and incubated for another 1.5 h. Then the chromogen was added and the plate was read at 450 nm. The fibronectin protein was quantified by competitive sandwich ELISA. Briefly, the samples were diluted and applied to the plate coated with the anti-fibronectin antibody, and the same amount of biotin-labeled fibronectin was immediately added to the wells. After incubation for 1.5 h and extensive washing, the horseradish peroxidase-conjugated streptavidin was added to the wells and incubated for 30 min. Then the chromogen was added and the plate was read at 450 nm.
Statistical analysis All data were presented as mean±SD. Statistical analysis was performed by one-way ANOVA using SPSS 11.0 (SPSS, Chicago, IL, USA). P<0.05 was considered statistically significant.
Results
Changes of basic parameters Blood glucose levels of both the DM and DM+Flu groups maintained at a high level of >16.7 mmol/L during the 4 weeks. Treatment with fluva-statin had no effect on the elevated blood glucose level. There was no significant difference in systolic blood pressure, total cholesterol, and triglyceride levels among the 3 groups. Urinary albumin excretion (UAE)was about 2.5-fold higher in the DM group than that of the non-DM group, and fluvastatin treatment significantly alleviated proteinuria. The kidney/body weight ratio was increased in DM group compared with that of the non-DM group; the treatment with fluvastatin was associated with a significant reduction in the kidney/body weight ratio. In addition, we measured the glomerular diameter (MGD). The MGD increased in the DM group compared with that of the non-DM group (P<0.01); fluvastatin treatment significantly inhibited glomerular hypertrophy (P<0.05; Table 1).
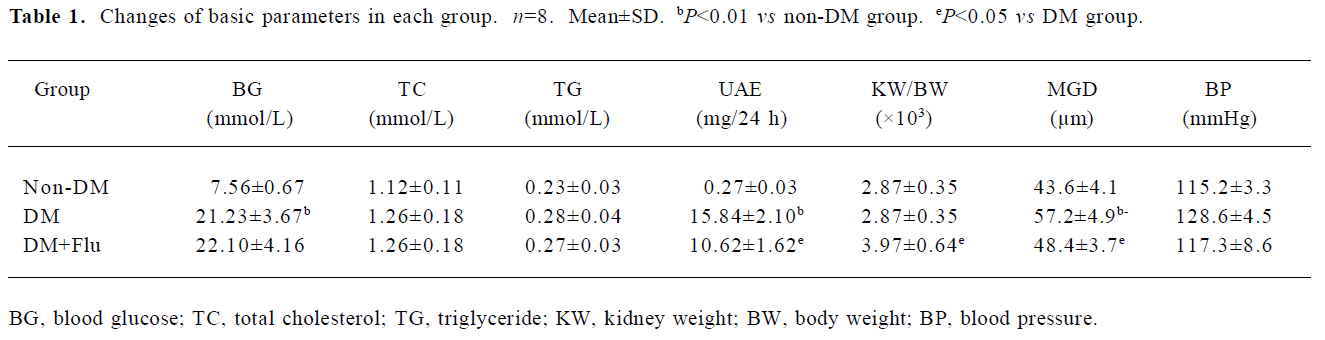
Full table
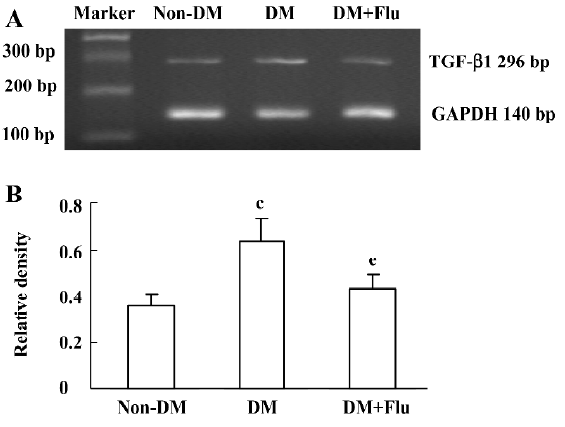
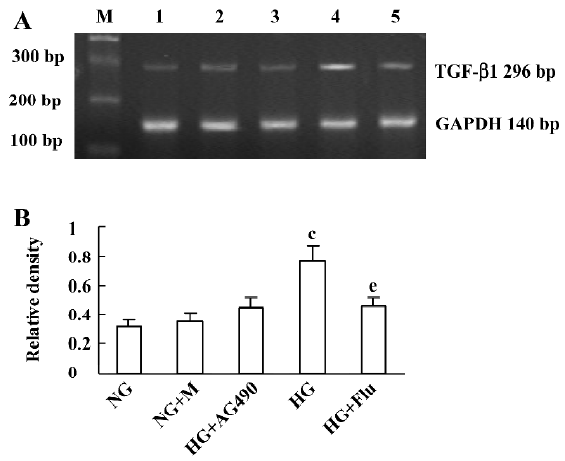
Effect of fluvastatin on the transcription of TGF-β1 mRNA The RT-PCR analysis indicated that the mRNA level of TGF-β1 was increased significantly in diabetic glomeruli when compared with that of non-diabetic glomeruli. The treatment of fluvastatin downregulated glomerular TGF-β1 mRNA expression in the diabetic kidney (Figure 1). We cultured GMC under NG or HG conditions and treated them with fluvastatin or AG490. TGF-β1 mRNA expression was upregulated by HG stimulation in the mesangial cells, and the treatment of AG490 or fluvastatin effectively inhibited the HG-induced upregulation of the TGF-β1 mRNA level (Figure 2).
Effect of fluvastatin on the production of TGF-β1 and fibronectin We examined the concentration of the TGF-β1 protein and fibronectin in the culture medium of the GMC with ELISA analysis. We found that the GMC exposed to HG for 48 h showed higher levels of the TGF-β1 protein and fibronectin than those cultured under NG condition. When GMC were treated with AG490 or fluvastatin, the secretion of TGF-β1 and fibronectin decreased (Table 2).
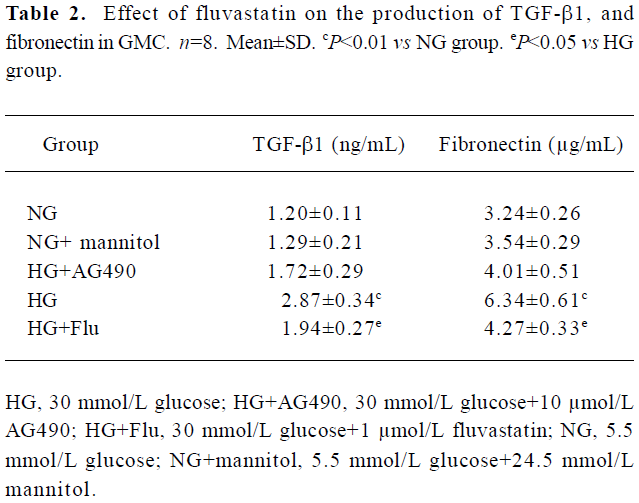
Full table
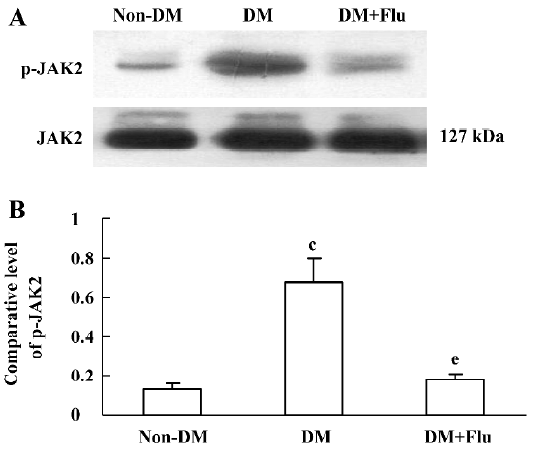
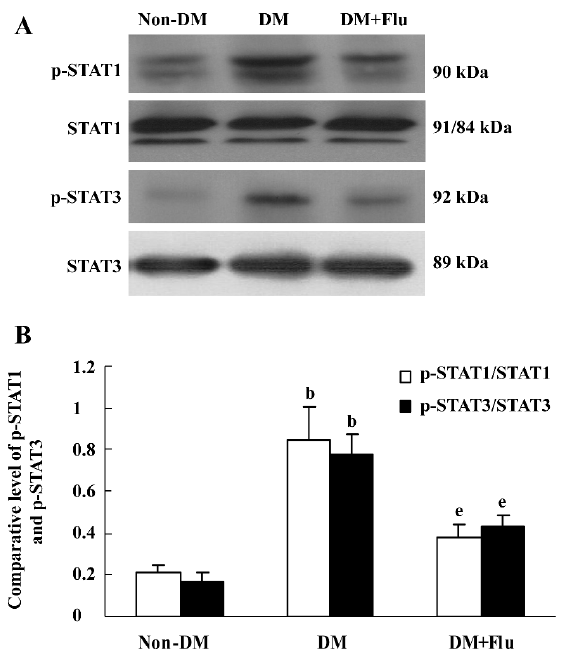
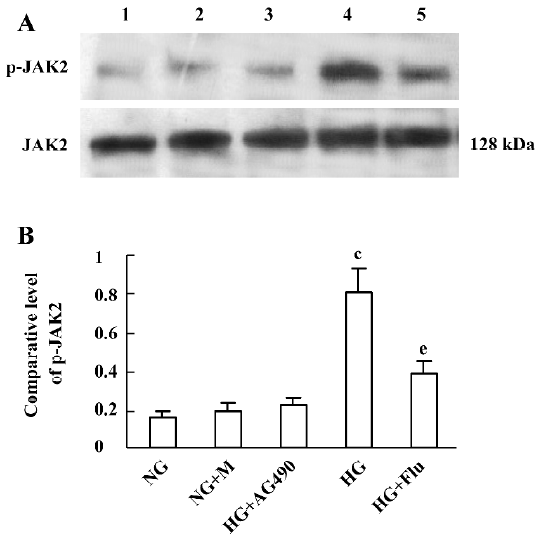
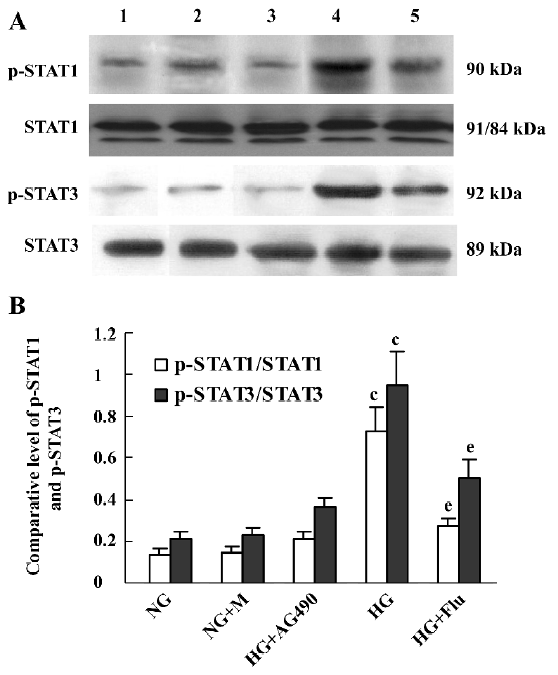
Effect of fluvastatin on the activation of the JAK2 and STAT proteins To determine the effect of fluvastatin on the activation of the JAK2 and STAT proteins, we first examined the phosphorylation of JAK2, STAT1, and STAT3 in diabetic glomeruli by Western blot analysis. The result demonstrated that the expression levels of tyrosine phosphorylation of JAK2, STAT1, and STAT3 obviously increased in the DM group compared with the non-DM group. Fluvastatin treatment significantly reduced glomerular tyrosine phosphorylation of JAK2, STAT1, and STAT3 in diabetic kidneys (P<0.05, Figures 3, 4). We further confirmed the effect of fluvastatin on the activation of JAK2 and STAT proteins using GMC cultured under HG condition. In accordance with the in vivo data, the expression levels of the tyrosine phosphorylation of JAK2, STAT1, and STAT3 significantly increased under HG conditions, compared with of the NG and NG+mannitol groups (P<0.01). The JAK2 inhibitor AG490 significantly inhibited the phosphorylation of JAK2, STAT1, and STAT3 in GMC cultured in medium with HG (P<0.05). Meanwhile, the tyrosine phosphorylation of JAK2, STAT1, and STAT3 significantly reduced (P<0.05) with the addition of fluvastatin to the culture medium (Figures 5, 6).
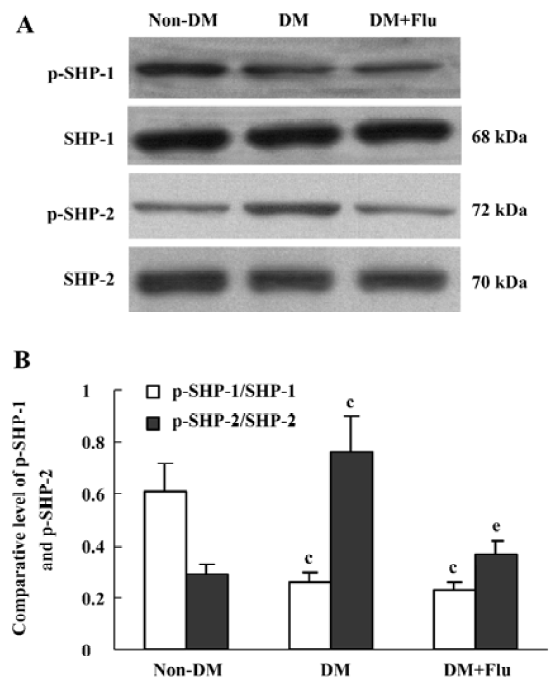
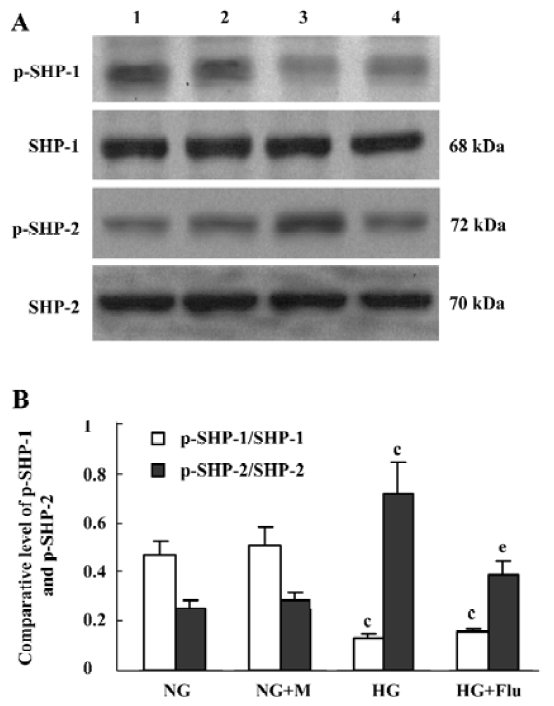
Effect of fluvastatin on the activation of SHP-1 and SHP-2 The expression ratio of tyrosine phosphorylated SHP-1/total SHP-1 significantly decreased in DM group compared with the non-DM group; whereas, SHP-2 tyrosine phosphorylation increased in the DM group. Fluvastatin significantly inhibited the tyrosine phosphorylation of SHP-2, but no significant effect was seen on the dephosphorylation of SHP-1 (Figure 7). We also investigated the effects of fluvastatin on the activation of SHP-1 and SHP-2 in GMC by examining their tyrosine phosphorylation levels. The ratio of phosphorylated SHP-1/total SHP-1 was lower in the HG group than in the NG and NG+M groups; whereas the GMC in HG group showed increased SHP-2 phosphorylation. Fluvastatin significantly inhibited the HG-induced tyrosine phosphorylation of SHP-2, but it had no effect on SHP-1 phosphorylation in GMC under HG conditions (Figure 8).
Discussion
Statins are lipid-lowering agents that specifically, competitively, and reversibly inhibit the HMG-CoA reductase, the enzyme that catalyzes the conversion of HMG-CoA to mevalonic acid, which is the rate-limiting step in the formation of cholesterol. Cardiovascular benefits of statins have been conventionally attributed to the reduction in levels of low-density lipoprotein cholesterol. More recently, subanalyses of large clinical trials suggest that statins may also prove beneficial in ameliorating the progression of kidney disease through their cholesterol-dependent and/or cholesterol-independent effects[17]. In the hypertensive model of diabetic nephropathy, cerivastatin decreased albuminuria through the suppression of glomerular hyperfiltration, mesangial expansion, and the loss of charge barrier independently of a cholesterol-lowering effect[18].
In the present study, untreated STZ-induced diabetic rats exhibited significantly increased urinary albumin excretion (UAE), kidney weight/body weight ratio, and mean glomerular diameter. Fluvastatin treatment resulted in the amelioration of albuminuria and glomerular hypertrophy, without altering serum total cholesterol and triglyceride levels. Our findings suggest that these renoprotective effects are independent of the cholesterol-lowering effect.
Diabetic nephropathy is characterized by excessive deposition of ECM in the kidney, leading to glomerular mesangial expansion and tubulointerstitial fibrosis. Recent studies have demonstrated that early renal hypertrophy is detrimental to the kidney in the long term and is a precursor of development of renal fibrosis[19,20]. Although the exact mechanisms of renal hypertrophy are still unclear, several growth factors, cytokines, chemokines, and vasoactive agents have been implicated in the development of renal hypertrophy, which include TGF-β, insulin like growth factor-1 (IGF-1), and platelet derived growth factor (PDGF)[21–23]. Among them, TGF-β is an effecter molecule studied extensively as a major mediator of the hypertrophic and prosclerotic changes in diabetic kidney disease[24]. Our result demonstrated that the transcription level of TGF-β1 was increased in diabetic glomeruli and this increase was suppressed by fluvastatin. To confirm the fluvastatin effect on the glomerular TGF-β1 expression in diabetic rats, cultured rat GMC were also examined. Our study showed that GMC cultured under HG conditions produce TGF-β1 and ECM fibronectin at a significantly faster rate than those cultured under NG conditions, and the transcription level of TGF-β1 increased in the HG group. After treatment with fluvastatin, the transcription level of TGF-β1 and the secretion of TGF-β1 and fibronectin were decreased. This result suggests that fluvastatin may prevent diabetic nephropathy by the suppression of glomerular TGF-β1 expression.
The JAK/STAT pathway is an important link between cell surface receptors and nuclear transcriptional events leading to cell growth[3]. The JAK/STAT pathway, especially the JAK2-STAT1-dependent pathway, contributes to HG-induced overproduction of TGF-β and fibronectin in GMC[4]. Treatment with JAK2 inhibitor AG490 lowered systolic blood pressure and significantly reduced urinary protein excretion in diabetic rats[9]. In our present study, the STZ-induced diabetic rats revealed increased phosphorylation of JAK2, STAT1, and STAT3, and fluvastatin resulted in the inhibition of JAK2, STAT1, and STAT3 phosphorylation. Our result also demonstrated that fluvastatin significantly inhibited the increased phosphorylation of JAK2, STAT1, and STAT3 in GMC cultured under HG conditions. Meanwhile, AG490 blocked HG-induced JAK2, STAT1, and STAT3 phosphorylation and production of TGF-β1 and fibronectin. These results suggest that the renoprotective effects of HMG-CoA reductase inhibitor fluvastatin may be partly through the inhibitory activation of the JAK/STAT signaling pathway.
SHP-1 and SHP-2 are 2 Src homology 2 domain-containing tyrosine phosphatases with major pathological implications in cell growth-regulating signaling. The phosphorylation state of JAK2 is tightly regulated by SHP-1 and SHP-2 in GMC[7]. SHP-1 and SHP-2 have opposite roles in Ang II-induced JAK2 phosphorylation[25]. SHP-1 appears to act as a conventional phosphatase, promoting JAK2 dephosphorylation and the termination of the Ang II-induced JAK/STAT signaling[25]. On the other hand, SHP-2 seems to play an essential role in promoting JAK2 phosphorylation and the initiation of the Ang II-induced JAK/STAT cascade, leading to cell proliferation[25]. In this study, we investigated the phosphorylation of 2 cytosolic tyrosine phosphatases, SHP-1 and SHP-2, in diabetic rat glomeruli and GMC under HG condition. Our results demonstrated that SHP-1 phosphorylation was decreased and SHP-2 phosphorylation was increased under hyperglycemic conditions. These findings are consistent with a previous study[9]. These results provide further support for the hypothesis that sustained JAK2 activation under hyperglycemic conditions might be partly due to decreased SHP-1 and increased SHP-2 phosphoryla-tion. We also found that fluvastatin significantly reduced the hyperglycemia-induced tyrosine phosphorylation of SHP-2. This result suggested that the inhibitory effect of fluvastatin on JAK2 phosphorylation might be partly due to decreased SHP-2 phosphorylation.
The mechanism by which HG promotes JAK2 activation may relate to an interaction of JAK2 with reactive oxygen species (ROS) induced by HG[4]. Recent studies have shown that ROS regulates the activity of the protein-tyrosine phosphatases SHP-1 and SHP-2[26,27]. Amiri et al[8] demonstrated that HG augmented Ang II-induced ROS production, VSMC proliferation, and tyrosine phosphorylation of JAK2 via the polyol pathway activation of protein kinase C-β2, which in turn activated NADPH oxidase to produce ROS. ROS regulate the activity of the protein tyrosine phosphatases SHP-1 and SHP-2, and SHP-1 and SHP-2 in turn regulated the activation of JAK2. HG, advanced glycation end products, Ang II, and TGF-β1 all increase intracellular ROS in renal cells and contribute to the development and progression of diabetic renal injury[28]. Recent studies have demonstrated that statins may prevent the production of ROS[29,30]. Fluvastatin could increase renal intrinsic antioxidant enzyme activities and improve renal function in STZ-induced diabetic rats[11]. Therefore, the fluvastatin-regulated activation of the JAK/STAT pathway may be partly via reducing ROS production in GMC and diabetic kidneys.
In VSMC, Ang II has been shown to stimulate tyrosine phosphorylation and the activation of SHP-2, and HG appears to enhance the effects of Ang II on both SHP-2 tyrosine phosphorylation and SHP-2 activity[31]. Ang II type 1 (AT1)-receptor antagonist candesartan significantly reduced the hyperglycemia-induced tyrosine phosphorylation of SHP-2 in diabetic glomeruli[9]. This result demonstrated that tyrosine phosphorylation of SHP-2 was probably via the AT1 receptor. A previous study suggested that SHP-2 functions as an adaptor protein for the JAK2 association with the AT1 receptor[25]. Recent evidence revealed that treatment with statins decreased AT1 receptor expression in VSMC in vitro and in vivo. Fluvastatin reduced AT1 receptor protein and mRNA expression with a time-course in VSMC[30,32]. There-fore, the possible mechanism by which fluvastatin regulated SHP-2 phosphorylation was partly due to decreasing AT1 receptor expression.
In summary, our data demonstrate that fluvastatin exerts an inhibitory effect on the activation of the JAK/STAT pathway in diabetic rat glomeruli and GMC under HG conditions. The results provide further evidence that inhibiting JAK/STAT signaling might be an effective therapeutic strategy for diabetic nephropathy.
References
- Kim SI, Han DC, Lee HB. Lovastatin inhibits transforming growth factor-β1 expression in diabetic rat glomeruli and cultured rat mesangial cells. J Am Soc Nephrol 2000;11:80-7.
- Oh JH, Ha H, Yu MR, Lee HB. Sequential effects of high glucose on mesangial cell transforming growth factor-β1 and fibronectin synthesis. Kidney Int 1998;54:1872-8.
- Marrero MB, Banes AK, Stern DM, Eaton DC. Role of the JAK/STAT signaling pathway in diabetic nephropathy. Am J Physiol Renal Physiol 2006;290:F762-8.
- Wang X, Shaw S, Amiri F, Eaton DC, Marrero MB. Inhibition of the Jak/STAT signaling pathway prevents the high glucose-induced increase in tgf-beta and fibronection synthesis in mesangial cells. Diabetes 2002;51:3505-9.
- Aaronson DS, Horvath CM. A road map for those who don’t know JAK- STAT. Science 2002;296:1653-5.
- Darnell JE, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to INFs and other extracellular signaling proteins. Science 1994;264:1415-21.
- Amiri F, Shaw S, Wang X, Tang J, Waller JL, Eaton DC, et al. Angiotensin II activation of the JAK/STAT pathway in mesangial cells is altered by high glucose. Kidney Int 2002;61:1605-16.
- Amiri F, Venema VJ, Wang X, Ju H, Venema RC, Marrero MB. Hyperglycemia enhances angiotensin II-induced janus-activated kinase/STAT signaling in vascular smooth muscle cells. J Biol Chem 1999;274:32382-6.
- Banes AK, Shaw S, Jenkins J, Redd H, Amiri F, Pollock DM, et al. Angiotensin II blockade prevents hyperglycemia-induced activation of JAK and STAT proteins in diabetic rat kidney glomeruli. Am J Physiol Renal Physiol 2004;286:F653-9.
- Oda H, Keane WF. Recent advances in statins and the kidney. Kidney Int Suppl 1999;71:2-5.
- Kurusu A, Shou I, Nakamura S, Fukui M, Shirato I, Tomino Y. Effects of the new hydroxy-3-methylglutaryl coenzyme A reductase inhibitor fluvastatin on anti-oxidant enzyme activities and renal function in streptozotocin-induced diabetic rats. Clin Exp Pharmacol Physiol 2000;27:767-70.
- Shibata S, Nagase M, Fujita T. Fluvastatin ameliorates podocyte injury in proteinuric rats via modulation of excessive Rho signaling. J Am Soc Nephrol 2006;17:754-64.
- Horiuchi M, Cui T, Li Z, Li J, Nakagami H, Iwai M. Fluvastatin enhances the inhibitory effects of a selective angiotensin II type 1 receptor blocker, vasartan, on vascular neointimal formation. Circulation 2003;107:106-12.
- Banes AK, Shaw S, Ma G, Brands M, Eaton DC, Stern DM, et al. Effect of simvastatin on high glucose- and angiotensin II-induced activation of the JAK/STAT pathway in mesangial cells. Am J Physiol Renal Physiol 2006;291:F116-21.
- Amiri F, Garcia R. Renal angiotensin II receptors and protein kinase C in diabetic rats: effects of insulin and ACE inhibition. Am J Physiol Renal Physiol 2000;278:F603-12.
- Amiri F, Garcia R. Regulation of angiotensin II receptors and PKC isoforms by glucose in rat mesangial cells. Am J Physiol 1999;276:F691-9.
- Shah S, Paparello J, Danesh FR. Effects of statin therapy on the progression of chronic kidney disease. Adv Chronic Kidney Dis 2005;12:187-95.
- Ota T, Takamura T, Ando H, Nohara E, Yamashita H, Kobayashi K. Preventive effect of cerivastatin on diabetic nephropathy through suppression of glomerular macrophage recruitment in a rat model. Diabetologia 2003;46:843-51.
- Wolf G. Vasoactive factors and tubulointerstitial injury. Kidney Blood Press Res 1999;22:62-70.
- Hannken T, Schroeder R, Stahl RAK, Wolf G. Atrial natriuretic peptide attenuates Ang II induced hypertrophy of renal tubular cells. Am J Physiol 2001;281:F81-90.
- Ling H, Vamvakas S, Schaefer L, Schnittler HJ, Schaefer RM, Heidland A. Angiotensin II induced cell hypertrophy: potential role of impaired proteolytic activity in cultured LLcpk1 cells. Nephrol Dialysis Transplant 1995;10:1305-12.
- Guo XH, Liu ZH, Dai CS, Li H, Liu D, Li LS. Rhein inhibits renal tubular epithelial cell hypertrophy and extracellular matrix accumulation induced by transforming growth factor β. Acta Pharmacol Sin 2001;22:934-8.
- Park IS, Kiyomoto H, Abboud SL, Abboud HE. Expression of transforming growth factor β and type IV collagen in early streptozotocin-induced diabetes. Diabetes 1997;46:473-80.
- Ziyadeh FN. Mediators of diabetic renal disease: the case for TGF-β as the major mediator. J Am Soc Nephrol 2004;15:S55-7.
- Marrero MB, Venema VJ, Ju H, Eaton DC, Venema RC. Regulation of angiotensin II-induced JAK2 tyrosine phosphorylation: roles of SHP-1 and SHP-2. Am J Physiol Cell Physiol 1998;275:C1216-23.
- Meng TC, Fukada T, Tonks NK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell 2002;9:387-99.
- Denu JM, Tanner KG. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry 1998;37:5633-42.
- Ha H, Lee HB. Reactive oxygen species and matrix remodeling in diabetic kidney. J Am Soc Nephrol 2003;14:S246-S249.
- Schaefer CA, Kuhlmann CR, Weiterer S, Fehsecke A, Abdallah Y, Schaefer C, et al. Statins inhibit hypoxia-induced endothelial proliferation by preventing calcium-induced ROS formation. Atherosclerosis 2006;185:290-6.
- Wassmann S, Laufs U, Baumer AT, Muller K, Konkol C, Sauer H, et al. Inhibition of geranylgeranylation reduces angiotensin II-mediated free radical production in vascular smooth muscle cells: involvement of angiotensin AT1 receptor expression and Rac1 GTPase. Mol Pharmacol 2001;59:646-54.
- Ali MS, Schieffer B, Delafontaine P, Bernstein KE, Ling BN, Marrero MB. Angiotensin II stimulates tyrosine phosphorylation and activation of insulin receptor substrate 1 and protein-tyrosine phosphatase 1D in vascular smooth muscle cells. J Biol Chem 1997;272:12373-9.
- Ichiki T, Takeda K, Tokunou T, Iino N, Egashira K, Shimokawa H, et al. Downregulation of angiotensin II type 1 receptor by hydrophobic 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 2001;21:1896-901.