
Neuroprotection by sodium ferulate against glutamate-induced apoptosis is mediated by ERK and PI3 kinase pathways1
Introduction
Glutamate is an excitotoxic neurotransmitter in the central nervous system. Its effects are mediated through the activation of glutamate receptors, which can be either coupled to G proteins or ionic channels that open upon binding of glutamate[1]. Of the latter, the N-methyl-D-aspartate (NMDA) receptors have been described as being associated with several pathological changes involving excessive stimulation of glutamate receptors[2]. A deregulated increase in the intracellular Ca2+ concentration caused by the overactivation of glutamate receptors is considered to trigger neuronal death through the activation of Ca2+-activated proteases, such as calpains or by indirectly activating the apoptotic-related caspases[3]. Glutamate neurotoxicity is known to be associated with numerous neurodegenerative disorders, including Alzheimer’s disease (AD), and is considered to be a key factor in the pathogenesis of AD[4]. Some studies have shown that amyloid β protein (Aβ) inhibits glutamate uptake and causes an increase in extracellular glutamate[5]. There are also some reports that Aβ enhances the toxicity induced by excitoxicity[6], indicating that Aβ-induced cytotoxicity might be mediated through glutamate cytoxicity to some extent. It has been shown that in vitro, glutamate induces different types of neuronal disorders, including apoptosis and necrosis. Therefore, the potential role of apoptosis in glutamate-induced toxicity suggests that its regulation may slow acute and chronic neurodegenerative processes.
Sodium ferulate (SF), extracted from a traditional Chinese herbal medicine, has potent antioxidant[7] and anti-inflammatory activities[8]. It has recently been reported that the long-term administration of ferulic acid protects mice from learning and memory deficits induced by centrally administered β-amyloid[9]. The primary action site of ferulic acid could be the microglia[10] and astrocytes[11]. A recent report showed that ferulic acid inhibited the formation of Aβ fibrils and destabilized preformed fibrillary Aβ[12]. Sultana et al reported that ferulic acid ethyl ester significantly inhibited Aβ1-42-induced cytoxicity, intracellular reactive oxygen species accumulation, lipid peroxidation, and the induction of induci-ble nitric oxide synthase (iNOS) in primary hippocampal cultures[13]. In addition, ferulic acid attenuates iron-induced oxidative damage and apoptosis in cultured neurons[14] and reduces the expression of inducible NOS and cyclooxygenase activity following exposure to lipopolysaccharides[15]. Our previous study showed that SF had protective effects from Aβ-induced neurotoxicity through the suppression of p38 mitogen-activated protein kinase and the upregulation of extracellular signal-regulated kinase (ERK) and Akt[16,17]. In the present study, we investigated the roles of the ERK pathway and phosphatidylinositol 3-kinase (PI3K) pathway in the neuroprotection by SF from glutamate toxicity.
Materials and methods
Materials SF, a colorless powder with >99% purity, was obtained from Suzhou Changtong Chemical Co (Suzhou, China). Glutamate (Sigma Chemical Co, St Louis, MO, USA) and SF was dissolved with Dulbecco’s modified Eagle’s medium (DMEM, Sigma Chemical Co, USA). Phospho-mitogen-activated protein kinase kinase (MEK)1/2 (Ser217/221, N
Cell cultures Primary cultures were obtained from the cerebral cortex of 0–24 h-old Sprague–Dawley rats according to the procedures described previously[18]. Briefly, after removal of the meninges and white matter, the brain cortex was collected and resuspended in Hanks’ solution without Ca2+ and Mg2+ (D-Hanks). The cortex was then mechanically fragmented, transferred to D-Hanks’ solution containing 0.125% trypsin, and incubated for 15 min at 37 ℃. Following trypsinization, the cells were washed twice with DMEM containing 10% heat-inactivated fetal bovine serum and resuspended in neurobasal medium supplemented with 2% B27 supplement, 5% heat-inactivated horse serum, 50 U/mL penicillin, and 50 mg/mL streptomycin. Aliquots of 1×106 cells/mL were seeded onto tissue culture plates precoated with poly-L-lysine (0.1 g/L) and kept at 37 ℃ in a humidified atmosphere with 5% CO2 and 95% O2. Forty eight hours after plating, cytosine arabinofuranoside was added to maintain a final concentration of 5 µmol/L to inhibit the proliferation of non-neuronal cells. At d 3, one-half of the medium was removed and replaced by the same volume of fresh medium. Only mature cultures (10–13 d in vitro) were used for the experiments. Our preliminary experiments using microtubule-associated protein immunostaining and glial fibrillary acidic protein (GFAP) immunostaining indicated that about 95% of the cells were neurons after arabinofuranoside treatment for 3 d.
Treatment of the cultures The rat neurons cultured for 10–13 d were incubated with 50 µmol/L glutamate for either 30 min or 24 h, with or without SF treatment (100, 200, and 500 µmol/L, respectively). In co-incubation experiments, SF was added to the neurons 30 min prior to incubation with glutamate. LY294002 or wortmannin (inhibitors of PI3K phosphorylation) and PD98059 or U0126 (inhibitors of MEK phosphorylation) were added to the cell cultures 1 h prior to SF treatment. The medium was gently removed at the indicated times, and the attached cells were fixed for the morphological assessment of apoptosis. In addition, total and cytosolic protein fractions were extracted for Western blotting.
Quantification of apoptosis by nuclear morphological changes Apoptosis was assessed by an analysis of nuclear morphology. Briefly, the neurons were fixed with 4% formaldehyde in phosphate-buffered saline (PBS, pH 7.4) for 10 min at room temperature, incubated with Hoechst 33258 dye at a concentration of 5 mg/L in PBS for 10 min in the dark, and then washed with PBS. Fluorescence was visualized using an Axioskop fluorescence microscope (Carl Zeiss GmbH, Jena, Germany). Fluorescent nuclei were scored and categorized according to the condensation and staining characteristics of chromatin. Normal nuclei showed non-condensed chromatin dispersed over the entire nucleus. Apoptotic nuclei were identified by condensed chromatin contiguous to the nuclear membrane and by nuclear fragmentation and apoptotic bodies. Both the apoptotic and the normal nuclei were counted under a fluorescent microscope with a magnification of 40× of the field lens. On each culture slide, the nuclei in 4 randomly selected microscopic fields were counted. For each group, the nuclei in the 4 slides were counted (altogether, the nuclei in 16 microscopic fields were counted in each group). The mean values were calculated and the data were expressed as the percentage of the apoptotic nuclei among the total nuclei.
Western blot analysis Western blot was performed to analyze the expression of phospho-MEK1/2, phospho-ERK1/2, phospho-Akt/ protein kinase B (PKB), phospho-p70S6K, caspase-3, m-calpains, PARP, cleaved PARP, and Bcl-2. The cells were washed once with ice-cold PBS, harvested, and centrifuged at 600×g for 10 min at 4 ℃. The pellets were lysed in ice-cold lysis buffer [1% Triton, 0.1% SDS, 0.5% deoxycholate, 1 mmol/L EDTA, 20 mmol/L Tris (pH 7.4), 150 mmol/L NaCl, 10 mmol/L NaF, and 0.1 mmol/L phenylmethyl-sulfonyl fluoride (PMSF)]. The nuclear fractions were first isolated by centrifuging the homogenates at 7500×g for 30 min at 4 ℃. The supernatant was further centrifuged at 12 000×g for 20 min at 4 ℃C to remove insoluble materials. The protein concentrations were quantified by the method of Lowry. The samples were equalized for protein concentrations. The proteins were separated with 10%–12% SDS-PAGE and transferred onto nitrocellulose membranes. The membranes were blocked with 3% bovine serum albumin in Tris-buffered saline (TBS, pH 7.6) for 1 h and incubated overnight at 4 ℃ with suitably diluted primary antibodies. After extensive washed with TBS-Tween (0.5% Tween 20), the membranes were incubated with antirabbit IgG, a HRP-linked antibody, and an antibiotin antibody for 1 h at room temperature. The blots were detected using the enhanced chemiluminescence (ECL) reaction. After visualization by ECL, all of the nitrocellulose strips were reprobed with the β-actin antibody to ensure equal loading of the proteins on all SDS-PAGE gels. The immunoreactive blots were incubated with the alkaline phosphatase-conjugated antimouse IgG antibody for 1 h. Finally, the blots were developed with the alkaline phosphatase substrate O-dianisidine tetrazotized along with β-naphthyl acid phosphate. Quantification of protein bands was achieved by densitometric analysis using Chemimage 5500 software (UVP, Pittsburgh, PA, USA).
Statistical analysis All data were presented as mean±SD. Statistical analysis was carried out with one-way ANOVA, followed by least significant difference (LSD)’s post-hoc test, which was provided by SPSS 11.5 statistical software (SPSS, Chicago, IL, USA). The level of significance was accepted as P<0.05.
Results
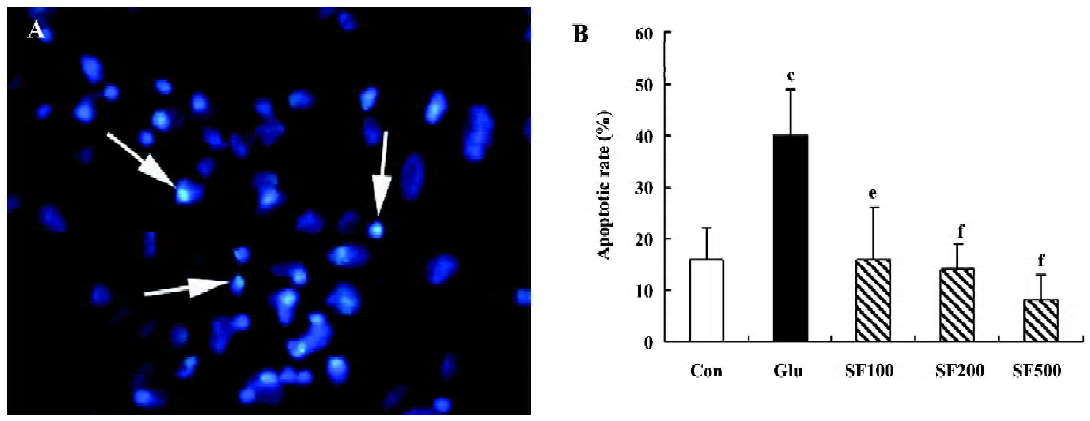
SF prevents glutamate-induced apoptosis in cultured cortical neurons Cultured cortical neurons were incubated with 50 µmol/L glutamate for 24 h and examined for the characteristic morphological nuclear changes associated with apoptosis. After 24 h of incubation, about 40%±9% of cells displayed apoptotic morphology, characterized by chromatin condensation. SF (100, 200, and 500 µmol/L, respectively) significantly reduced the number of apoptotic cells induced by glutamate in a dose-dependent manner (Figure 1).
The activation of caspase-3 is a hallmark of apoptosis and precedes changes in nuclear morphology. The activated caspase-3 of 20 kDa was observed with the Western blot analysis. The results showed a remarkable increase of the active caspase-3 fragment in the neurons incubated with glutamate compared with the controls. The pretreatment with SF almost reduced the glutamate-induced increase in activated caspase-3 compared to the control values (Figure 2A, 2B).
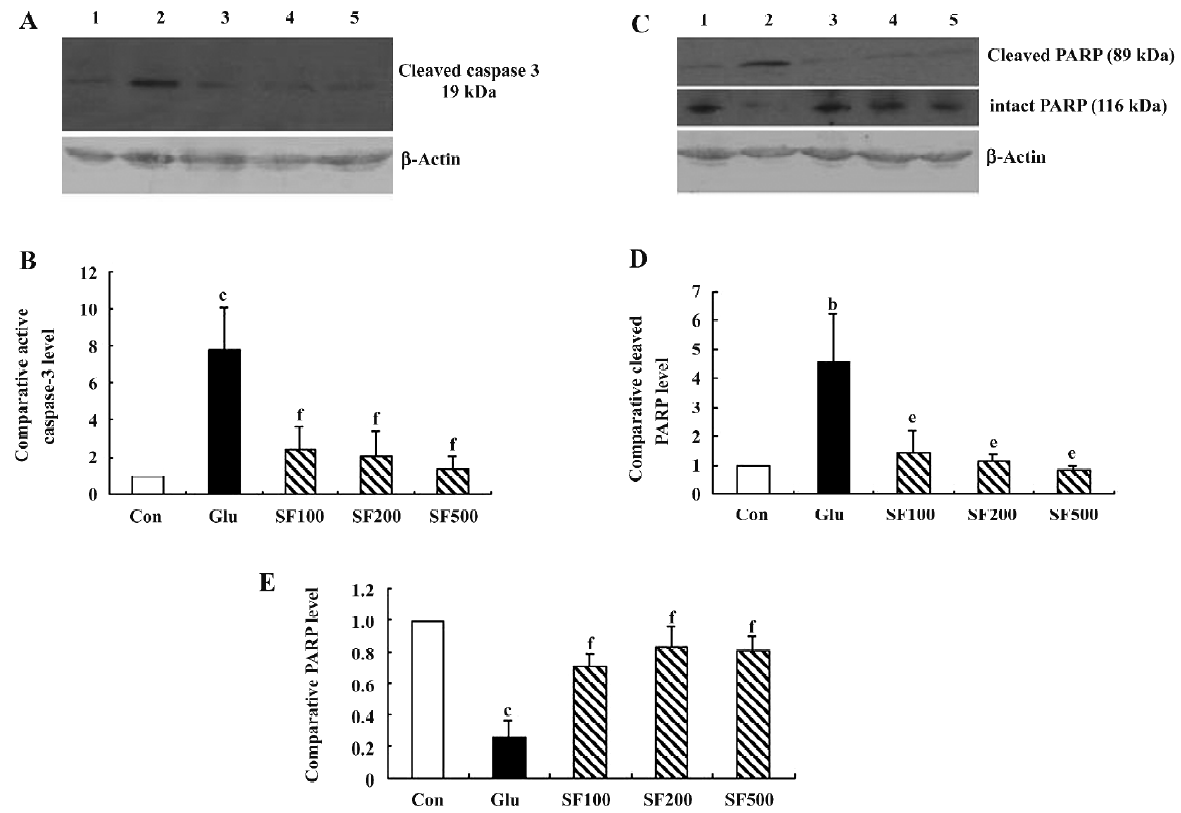
During apoptosis, poly (ADP-ribose) PARP is one of the earliest targets for caspase-3 cleavage which induces the formation of a 89 kDa C-terminal fragment containing the catalytic domain and a 24 kDa fragment that binds DNA ends[19]. As shown in Figure 2C, the level of intact PARP (116 kDa) was significantly decreased in the cortical neurons incubated with glutamate and was associated with the appearance of the 89 kDa fragment of PARP. In SF-pretreated neurons, the expression level of the intact PARP (116 kDa) was up-regulated while the expression of the 89 kDa fragment was lower than those in the neurons incubated with glutamate alone (Figure 2C–2E).
Taken together, these finding suggest that glutamate-mediated apoptosis in the rat cortical neurons is markedly inhibited by SF.
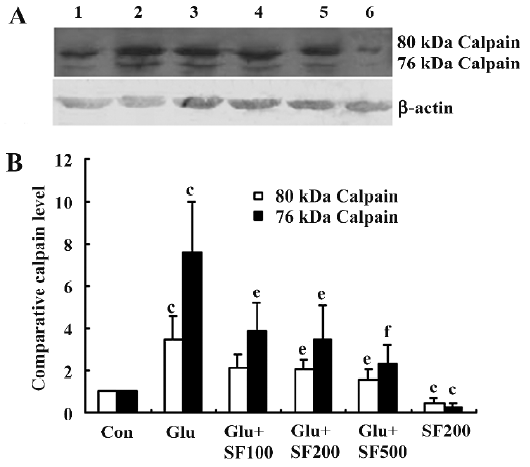
SF inhibits µ-calpain upregulation in cortical neurons exposed to glutamate The activation of calpains is now considered to play a key role in excitotoxic neuronal damage[20,21]. Therefore, in order to investigate whether SF prevents glutamate-induced µ-calpain activation, we used Western blotting to determine the activation of µ-calpain in rat primary cortical neurons after SF treatments. We found that compared with untreated cortical neurons, both 80 and 76 kDa µ-calpain bands were significantly increased in cortical neurons following exposure to glutamate for 24 h. Pretreatment of neurons with SF (100, 200, and 500 µmol/L, respec-tively) significantly reduced the glutamate-induced production of µ-calpain, both of the inactivated 80 kDa and activated 76 kDa forms in a concentration-dependent manner. Furthermore, SF alone significantly downregulated the basal expression levels of µ-calpain (Figure 3).
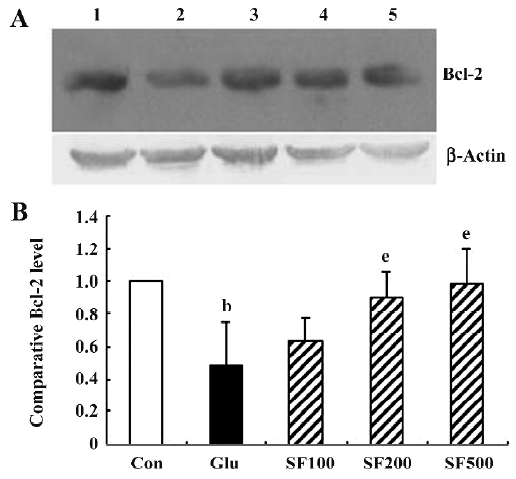
SF inhibits glutamate-induced downregulation of Bcl-2 protein levels in cultured cortical neurons The Bcl-2 protein plays a key role in the anti-apoptotic process. The incubation of neurons with glutamate for 30 min significantly reduced Bcl-2 protein levels as compared with the control. Pretreatment with SF (100, 200, and 500 µmol/L, respectively) significantly inhibited the reduction of Bcl-2 expression induced by glutamate in a dose-dependent manner (Figure 4), suggesting that Bcl-2 may mediate, to some extent, the protective effects provided by SF against glutamate toxicity. In addition, SF alone (500 µmol/L) had no obvious effect on the basal expression of Bcl-2 in the cultured cortical neurons (data not shown).
PI3K/Akt/p70S6K pathway is involved in neuroprotec-tion by SF against glutamate toxicity To investigate the role of the PI3K pathway in the protection by SF against glutamate toxicity, the neurons were pre-incubated with either LY294002, a synthetic bioflavonoid that reversibly binds to and inhibits p110[22] or wortmannin, a fungal toxin that covalently binds to and blocks the activity of the catalytic p110 subunit of PI3K[23]. As assessed by Hoechst 33258 staining, LY294002 (10 µmol/L) and wortmannin (100 nmol/L) significantly reduced the protective effects of SF against glutamate cytotoxicity (Figure 5A), suggesting that SF exerted an anti-apoptotic effect on cortical neurons exposed to excitotoxic insult through the PI3K/Akt pathway. A control experiment revealed that 2 PI3K inhibitors used had no effect on the toxicity of glutamate in the absence of SF. To test for possible toxic effects of those inhibitors, cortical neurons were treated with LY294002 and wortmannin alone at the concentration used in this study and no toxicity was observed (data not shown). We examined the level of phosphorylated Akt with hybridizing to the antibody specific against phospho-Akt (Thr308) using Western blotting. The basal level of phospho-Akt in cultured cortical neurons was relatively high. The treatment of neurons with glutamate for 30 min induced a significant decrease in the phosphorylated level of Akt. SF (500 µmol/L) prevented a glutamate-induced decrease of phosphorylated Akt. LY294002 (10 µmol/L) counteracted the effect of SF on phosphorylated Akt (Figure 5B,5C). In addition, SF treatment alone (500 µmol/L) exerted an increase in phosphorylated Akt expression that did not reach statistical significance compared with the untreated cortical neurons (data not shown).
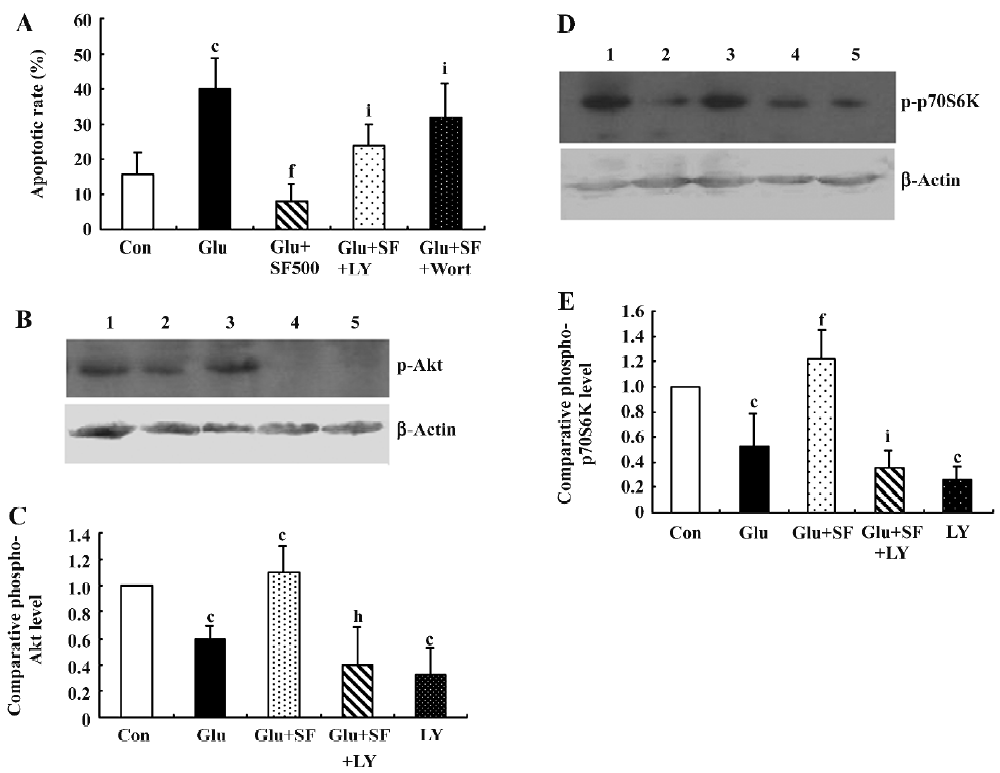
Because p70S6K acts downstream of Akt, we examined the level of phosphorylated p70S6K detected by an antibody specific for phospho-p70S6K (Thr389). The results showed that incubation of the neurons with glutamate for 30 min elicited a significant decrease in the phosphorylated level of p70S6K. SF (500 µmol/L) prevented a glutamate-induced decrease in the expression of phosphorylated p70S6K. This effect on the phosphorylation of p70S6K was blocked by the application of LY294002, indicating that the SF-induced phosphorylation of p70S6K depended on PI3K activation (Figure 5D,5E). Similarly, SF treatment alone did not elicit a significant increase in phosphorylated p70S6K expression (data not shown).
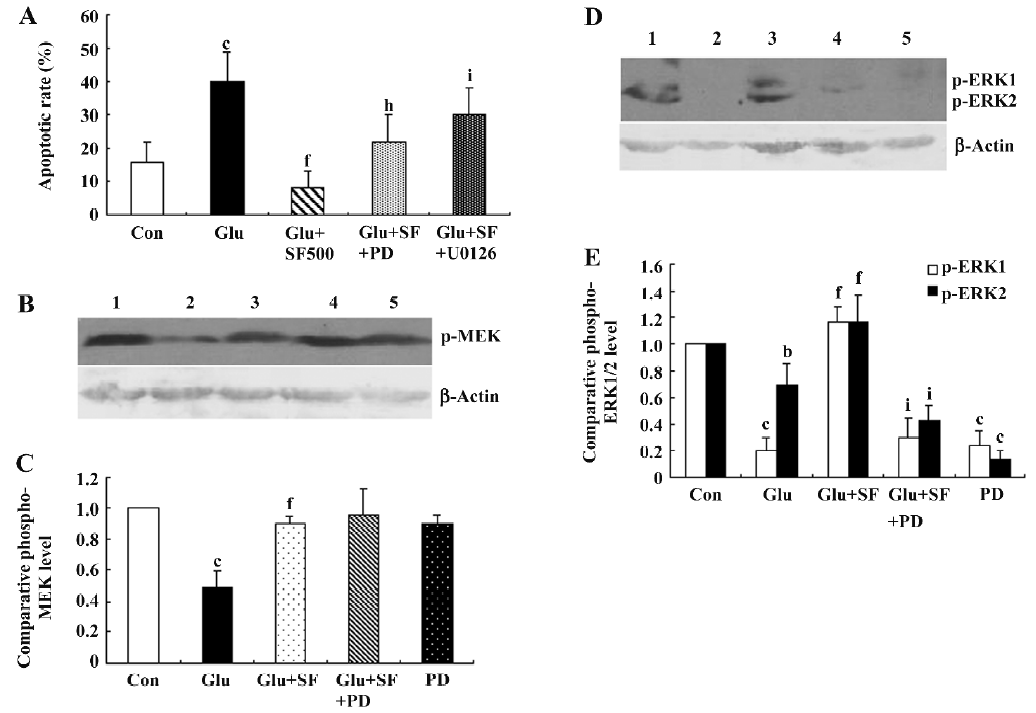
MEK/ERK pathway is involved in neuroprotection by SF against glutamate toxicity. In order to determine whether the activation of the ERK pathway was involved in the neuroprotective effect of SF, we pharmacologically inhibited MEK, the upstream kinase of ERK, with PD98059 (10 µmol/L) or U0126 (30 µmol/L)[24]. In the presence of PD98059 or U0126, the neuroprotective effect of SF against glutamate-evoked cell death was partially abrogated, as observed by an analysis of nuclear morphology, indicating that the ERK pathway partially mediates the neuroprotective effect of SF against glutamate toxicity (Figure 6A). Control experiments revealed that the 2 MEK inhibitors used had no effect on glutamate toxicity measured in the absence of SF. To investigate for possible toxic effects of those inhibi-tors, cortical neurons were treated independently with PD98059 or U0126 at the concentration used in this study and no toxicity was observed (data not shown). Then, we determined the effect of SF on the phosphorylation of ERK1/2 in cultured cortical neurons by Western blotting. The results showed that the treatment of neurons with glutamate for 30 min induced a significant decrease in phosphorylated ERK1/2 expression (Figure 6D,6E). However, pretreatment with SF prevented the glutamate-induced decrease in phosphorylated ERK1/2 expression. SF-induced ERK1/2 phosphorylation was significantly blocked by the simultaneous application of PD98059 (10 µmol/L), suggesting that MEK1/2 activation mediates the SF-induced increase in ERK1/2 phosphorylation (Figure 6D, 6E). In the extended analysis of the ERK pathway, phosphorylation of MEK1/2 was detected. The results showed that treatment of the neurons with glutamate also elicited a significant decrease in phospho-MEK1/2. SF inhibited the glutamate-induced decrease in phospho-MEK1. PD98059 (10 µmol/L) did not block the effect of SF on phospho-MEK1 expression (Figure 6B, 6C). SF treatment alone did not elicit significant effects on basal phospho-MEK and phospho-ERK1/2 expressions in cultured cortical neurons (data not shown).
Discussion
The cell death induced by glutamate is believed to be involved in neuronal loss associated with both acute and chronic neurodegenerative insults. Thus, the dissection of glutamate signal transduction may have clinical significance for neuroprotection. This study demonstrated that pre-incubation with SF protects cultured cortical neurons against glutamate toxicity. The activations of the PI3K/Akt/p70S6K pathway and the MEK/ERK1/2 pathway play important roles in the protective effect of SF against glutamate toxicity in cortical neurons. Furthermore, SF upregulated the expression of the anti-apoptotic protein Bcl-2 and prevented the upregulation of the µ-calpain protein in rat cortical neurons after treatment with glutamate, suggesting that the downregulation of µ-calpain may also account for the neuroprotective effect of SF.
Calpains represent a superfamily of Ca2+-activated cysteine-proteases, which are important mediators of apoptosis and necrosis. In the brain, the presence of µ-calpain was shown primarily in neurons, whereas µ-calpain is more prominent in glial cells[25]. The activation of µ-calpain in cortical neurons specifically linked to Ca2+ entry through the NMDA receptor[26]. A recent study strongly suggested that calpain plays a central role as a excitotoxic signal transduction cascade leading to DNA fragmentation[20]. The results indicated that calpain cleaved Bid and that truncated Bid then translocated to mitochondria and induced mitochondria membrane permeabilization and the release of DNA fragmentation factors resulting in neurodegeneration[20]. In addition, it has been shown that calpains can cleave Bcl-2, thereby contributing to the ischemia-induced decrease in Bcl-2 protein levels thereby triggering the intrinsic apoptotic pathway[27,28]. Our studies indicated that SF inhibited the upregulation of µ-calpain in cortical neurons exposed to glutamate. This inhibitory effect of SF on µ-calpain expression may be partly responsible for the neuroprotective effects against glutamate-neurotoxicity. One explanation for the inhibitory effect of SF on µ-calpain expression could be that SF is a novel competitive NMDA receptor antagonist[29]. It is known that apopto-sis observed in neurons under exposure to glutamate depends on the hyperactivation of NMDA receptor and hence excessive Ca2+ influx through NMDA receptor channels[30]. Dizocilpine (MK-801), a specific antagonist of the NMDA receptor, was shown to almost completely block cell death induced by glutamate[31].
The precise mechanism of the protective effect of SF through the activation of PI3K/Akt/p70S6K pathway is unclear, but a possible explanation is that the activated p70S6K phosphorylates Bad. The phosphorylated Bad appears to be the inactive moiety sequestered in the cytosol bound to 14-3-3, freeing Bcl-x or Bcl-2 to promote survival[32]. Another explanation is that PI3K/Akt induces the upregula-tion of the anti-apoptotic protein Bcl-2. The direct induction of Bcl-2 by activated PI3K/Akt has been described in some studies[33]. In the current study, pretreatment with SF significantly inhibited the decrease of phospho-Akt and Bcl-2 expression induced by glutamate. It has been reported that infection with the Bcl-2-expressing viral vector protected cortical cells from glutamate excitotoxicity[34]. The results indicate that SF protects cortical neurons from glutamate-induced toxicity by inducing the overexpression of Bcl-2 through PI3K/Akt activation.
In order to investigate whether the activation of the ERK pathway was involved in the neuroprotective effect of SF, we inhibited the upstream kinase of ERK (MEK) with PD98059 or U0126. In the presence of the MEK inhibitors, the neuro-protective effect of SF against glutamate-evoked cell apo-ptosis was partially abrogated, as observed by an analysis of nuclear morphology, indicating that the ERK pathway mediates the neuroprotective effect of SF against glutamate toxicity. In further support of a role for MEK/ERK1/2 in SF-induced neuroprotection, SF prevented the glutamate-induced decrease in phosphorylated level of MEK1/2 and ERK1/2. The effect of SF on ERK1/2 phosphorylation was MEK-dependent as the application of PD98059 prevented this effect, further confirming the role of the ERK pathway in the neuroprotection by SF. The activation of the ERK signaling pathway was reported to suppress the pro-apoptotic activity of the stress-activated c-Jun N-terminal kinase (JNK)/p38 protein kinase, thus protecting rat pheochromocytoma cells (PC-12) from nerve growth factor NGF withdrawal-induced cell death[35]. The MEK/ERK pathway interferes with apoptosis at the level of cytosolic caspase activation, downstream of the release of cytochrome c from mitochondria[36].
The fact that both PI3K and MEK/ERK contribute to the protection of cortical neurons by SF from glutamate toxicity may be explained by the activation of multiple lethal reactions during excitotoxic cell death[37,38]. The simultaneous contributions of PI3K and MEK/ERK to the protection of cortical neurons by SF from glutamate-evoked apoptotic death may also be due to the cross-talk between the 2 pathways. A recent study showed that 3-phospho-inositide-dependent protein kinase (PDK)1 directly binds and activates MEK, thereby contributing to the activation of this pathway[39]. PI3K is also responsible for maintaining constitutive ERK1/2 activity in different cell lines, in which basal PI3K and ERK activities are required to prevent cell death[40]. The activation of the transcription factor cyclic adenosine monophosphate response element-binding protein (CREB) has been reported as a consequence of ERK activation. CREB is not a direct substrate for ERK. Rather, ERK needs to phosphorylate and activate members of the p90 ribosomal S6 kinase family, which in turn may phosphorylate and activate CREB[41]. Interestingly, CREB has also been identified as a regulatory target for c-Akt[42], suggesting that CREB may function as a convergence point of 2 survival-promoting signaling pathways. CREB is a key molecule in neuronal survival.
In conclusion, this study demonstrated that PI3K/Akt/p70S6K and the MEK/ERK signaling pathway play important roles in the protective effect of SF against glutamate toxicity in cortical neurons. Two different signal transduction pathways, the PI3K/Akt and the MEK/ERK1/2 pathways, might directly or indirectly inhibit the activation of cell death mechanism. The results reported here suggest that agents that selectively target the PI3K and/or the MEK/ERK pathways may be clinically useful in the protection of neurons against glutamate toxicity.
References
- Nakanishi S. Molecular diversity of glutamate receptors and implications for brain function. Science 1992;258:597-603.
- Lynch DR, Guttmann RP. Excitotoxicity: perspectives based on N-methyl-D-aspartate receptor subtypes. J Pharmacol Exp Ther 2002;300:717-23.
- Chan SL, Mattson MP. Caspase and calpain substrates: roles in synaptic plasticity and cell death. J Neurosci Res 1999;58:167-90.
- Kihara T, Shimohama S, Sawada H, Honda K, Nakamizo T, Kanki R, et al. Protective effect of dopamine D2 agonists in cortical neurons via the phosphatidylinositol 3 kinase cascade. J Neurosci Res 2002;70:274-82.
- Harris ME, Wang Y, Pedigo NW Jr, Hensley K, Butterfield DA, Carney JM. Amyloid beta peptide (25–35) inhibits Na+-dependent glutamate uptake in rat hippocampal astrocyte cultures. J Neurochem 1996;67:277-86.
- Kihara T, Shimohama S, Sawada H, Honda K, Nakamizo T, Shibasaki H, . alpha 7 nicotinic receptor transduces signals to phosphatidylinositol 3-kinase to block Abeta-amyloid-induced neurotoxicity. J Biol Chem 2001; 276: 13 541–6.
- Scott BC, Butler J, Halliwell B, Aruoma OI. Evaluation of the antioxidant action of ferulic acid and catechins. Free Radic Res Commun 1993;19:241-53.
- Fernandez MA, Saenz MT, Garcia MD. Anti-inflammatory activity in rats and mice of phenolic acids isolated from Scrophu-laria frutescens. J Pharm Pharmacol 1998;50:1183-6.
- Yan JJ, Cho JY, Kim HS, Kim KL, Jung JS, Huh SO, et al. Protection against beta-amyloid peptide toxicity in vivo with log-term administration of ferulic acid. Br J Pharmacol 2001;133:89-96.
- Kim HS, Cho JY, Kim DH, Yan JJ, Lee HK, Suh HW, et al. Inhibitory effects of long-term administration of ferulic acid on microglial activation induced by intracerebroventricular injection of beta-amyloid peptide (1-42) in mice. Biol Pharm Bull 2004;27:120-1.
- Cho JY, Kim HS, Kim DH, Yan JJ, Suh HW, Song DK. Inhibitory effects of long-term administration of ferulic acid on astrocyte activation induced by intracerebroventricular injection of beta-amyloid peptide (1-42) in mice. Prog Neuropsychopharmacol Biol Psychiatry 2005;29:901-7.
- Ono K, Hirohata M, Yamada M. Ferulic acid destabilizes preformed beta-amyloid fibrils in vitro. Biochem Biophys Res Commun 2005;336:444-9.
- Sultana R, Ravagna A, Mohmmad-Abdul H, Calabrese V, Butterfield DA. Ferulic acid ethyl ester protects neurons against amyloid beta-peptide(1–42)-induced oxidative stress and neurotoxicity: relationship to antioxidant activity. J Neurochem 2005;92:749-58.
- Zhang Z, Wei T, Hou J, Li G, Yu S, Xin W. Iron-induced damage and apoptosis in cerebellar granule cells: attenuation by tetra-methyl-pyrazine and ferulic acid. Eur J Pharmacol 2003;467:41-7.
- Hosoda A, Ozaki Y, Kashiwada A, Mutoh M, Wakabayashi K, Mizuno K, et al. Syntheses of ferulic acid derivatives and their suppressive effects on cyclooxygenase-2 promoter activity. Bioorg Med Chem 2002;10:1189-96.
- Jin Y, Yan EZ, Fan Y, Zong ZH, Qi ZM, Li Z. Sodium ferulate prevents amyloid-beta-induced neurotoxicity through suppression of p38 MAPK and upregulation of ERK-1/2 and Akt/protein kinase B in rat hippocampus. Acta Pharmacol Sin 2005;26:943-51.
- Jin Y, Fan Y, Yan EZ, Liu Z, Zong ZH, Qi ZM. Effects of sodium ferulate on amyloid-beta-induced MKK3/MKK6-p38 MAPK-Hsp27 signal pathway and apoptosis in rat hippocampus. Acta Pharmacol Sin 2006;27:1309-16.
- Sribnick EA, Ray SK, Nowak MW, Li L, Banik NL. 17beta-estradiol attenuates glutamate-induced apoptosis and preserves electrophysiologic function in primary cortical neurons. J Neurosci Res 2004;76:688-96.
- Soldani C, Scovassi AI. Poly(ADP-ribose)polymerase-1 cleavage during apoptosis: an update. Apoptosis 2002;7:321-8.
- Takano J, Tomioka M, Tsubuki S, Higuchi M, Iwata N, Itohara S, . Calpain mediates excitotoxic DNA fragmentation via mitochondrial pathways in adult brains: evidence from calpastatin mutant mice. J Biol Chem 2005; 280: 16 175–84.
- Higuchi M, Tomioka M, Takano J, Shirotani K, Iwata N, Masumoto H, . Distinct mechanistic roles of calpain and caspase activation in neurodegeneration as revealed in mice overexpressing their specific inhibitors. J Biol Chem 2005; 280: 15 229–37.
- Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J Biol Chem 1994;269:5241-48.
- Powis G, Bonjouklian R, Berggren MM, Gallegos A, Abraham R, Ashendel C, et al. Wortmannin, a potent and selective inhibitor of phosphatidylinositol-3-kinase. Cancer Res 1994;54:2419-23.
- Iacovelli L, Bruno V, Salvatore L, Melchiorri D, Gradini R, Caricasole A, et al. Native group-III metabotropic glutamate receptors are coupled to the mitogen-activated protein kinase/phosphatidylinositol-3-kinase pathways. J Neurochem 2002;82:216-23.
- Hamakubo T, Kannagi R, Murachi T, Matus A. Distribution of calpains I and II in rat brain. J Neurosci 1986;6:3103-11.
- Hewitt KE, Lesiuk HJ, Tauskela JS, Morley P, Durkin JP. Selective coupling of mu-calpain activation with the NMDA receptor is independent of translocation and autolysis in primary cortical neurons. J Neurosci Res 1998;54:223-32.
- Gil-Parrado S, Fernandez-Montalvan A, Assfalg-Machleidt I, Popp O, Bestvater F, Holloschi A, Ionomycin-activated calpain triggers apoptosis. A probable role for Bcl-2 family members. J Biol Chem 2002; 277: 27 217–26.
- Rami A. Ischemic neuronal death in the rat hippocampus: the calpain-calpastatin-caspase hypothesis. Neurobiol Dis 2003;13:75-88.
- Yu L, Zhang Y, Ma R, Bao L, Fang J, Yu T. Potent protection of ferulic acid against excitotoxic effects of maternal intragastric administration of monosodium glutamate at a late stage of pregnancy on developing mouse fetal brain. Eur Neuropsycho-pharmacol 2006;16:170-7.
- Choi DW. Excitotoxic cell death. J Neurobiol 1992;23:1261-76.
- Du Y, Bales KR, Dodel RC, Hamilton-Byrd E, Horn JW, Czilli DL, Activation of a caspase 3-related cysteine protease is required for glutamate-mediated apoptosis of cultured cerebellar granule neurons. Proc Natl Acad Sci USA 1997; 94: 11 657–62.
- Harada H, Andersen JS, Mann M, Terada N, Korsmeyer SJ. p70S6 kinase signals cell survival as well growth, inactivating the pro-apoptotic molecule BAD. Proc Natl Acad Sci USA 2001;98:9666-70.
- Ahmed NN, Grimes HL, Bellacosa A, Chan TO, Tsichlis PN. Transduction of interleukin-2 antiapoptotic and proliferative signals via Akt protein kinase. Proc Natl Acad Sci USA 1997;94:3627-32.
- Jia WW, Wang Y, Qiang D, Tufaro F, Remington R, Cynader M. A bcl-2 expressing viral vector protects cortical neurons from excitotoxicity even when administered several hours after the toxic insult. Brain Res Mol Brain Res 1996;42:350-3.
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 1995;270:1326-31.
- Erhardt P, Schremser EJ, Cooper GM. B-Raf inhibits programmed cell death downstream of cytochrome c release from mitochondria by activating the MEK/Erk pathway. Mol Cell Biol 1999;19:5308-15.
- Mattson MP, Culmsee C, Yu ZF. Apoptotic and antiapoptotic mechanisms in stroke. Cell Tissue Res 2000;301:173-87.
- White BC, Sullivan JM, DeGracia DJ, O’Neil BJ, Neumar RW, Grossman LI, et al. Brain ischemia and reperfusion: molecular mechanisms of neuronal injury. J Neurol Sci 2000;179:1-33.
- Sato S, Fujita N, Tsuruo T. Involvement of 3-phosphoinositide-dependent protein kinase-1 in the MEK/MAPK signal transduction pathway. J Biol Chem 2004; 279: 33 759–67.
- Versteeg HH, Evertzen MW, van Deventer SJ, Peppelenbosch MP. The role of the phosphatidyllnositide-3-kinase in basal mitogen-activated protein kinase activity and cell survival. FEBS Lett 2000;465:69-73.
- Xing J, Ginty DD, Greenberg ME. Coupling of the RAS-MAPK pathway to gene activation by RSK2, a growth factor-regulated CREB kinase. Science 1996;273:959-63.
- Du K, Montminy M. CREB is a regulatory target for the protein kinase Akt/PKB. J Biol Chem 1998; 273: 32 377–9.