
Dual roles of NF-κB in cell survival and implications of NF-κB inhibitors in neuroprotective therapy1
Introduction
NF-κB is ubiquitously expressed in peripheral and brain cells and regulates the expression of a wide variety of genes involved in cell survival, growth, stress responses, and immune and inflammatory processes[1–3]. This factor was first described by Sen and Baltimore in 1986 as a NF, that when activated by agents, such as bacterial lipopolysaccharide, bound to a 10 bp sequence in the enhancer region of the gene encoding the κ light chain (κ) of antibody molecules in B cells (B)[4]. NF-κB family members have been implicated in the development of the nervous system and plasticity of synapses[5–7]. NF-κB is persistently activated in cancer, chronic inflammation, neurodegenerative diseases, stress, stroke, trauma, heart disease, and other disease conditions[8,9]. As NF-κB is an important regulator in programmed cell death[10], it has been speculated that NF-κB may play important roles in normal brain function and neurodegenerative disorders[11,12].
NF-κB biology
c-Rel/NF-κB family NF-κB is composed of 5 members of the c-Rel (Rel) family, including NF-κB1 (p50), NF-κB2 (p52), RelA (p65), RelB, and Rel. All the Rel proteins contain a conserved N-terminal region, called the Rel homology domain (RHD). The N-terminal part of the RHD contains the DNA-binding domain, whereas the dimerization domain is located from C-terminal region of the RHD[13]. Close to the C-terminal end of the RHD lies the nuclear localization signal (NLS), which is essential for the transport of active NF-κB complexes into the nucleus[14]. NF-κB family proteins are divided into 2 groups based on C-terminal sequences of the RHD. The members of group 1 include NF-κB proteins p105 and p100, which are precursors of p50 and p52. Limited proteolysis is required to produce p50 and p52. The second group (the Rel proteins) mainly includes c-Rel (and its retroviral homologue v-Rel), RelB, and RelA (p65)[15]. All vertebrate NF-κB proteins can form homodimers or heterodimers, except for RelB, which can only form hetero-dimers. These homodimers and heterodimers that exhibit differential binding specificities are p50/RelA, p50/c-Rel, p52/c-Rel, p65/c-Rel, RelA/RelA, p50/p50, p52/p52, RelB/p50 and RelB/p52[16]. The term NF-κB commonly refers specifically to a p50–RelA (p50/p65) heterodimer, which is the major Rel/NF-κB complex in most cells[14].
NF-κB dimers are sequestered in the cytoplasm by a class of inhibitor proteins, called IκB. In mammalian cells, the major regulatory IκB proteins are IκB-α, IκB-β, IκB-ε, and Bcl-3. The most common complex that is activated in mammalian cells appears to involve IκB-α, which binds to the p50/RelA heterodimer. IκB function as inhibitors through ankyrin repeats that interact with the RHD in NF-κB to mask the NLS and inhibit the nuclear translocation of NF-κB. The N-termini of these IκB proteins constitute a signal response domain, which is targeted for phosphorylation and ubiquitination by a variety of stimuli. The newly-synthesized IκB-α protein actively shuttles between the nucleus and the cytoplasm and both inhibit nuclear import and mediate the nuclear export of NF-κB/Rel proteins. In contrast, the IκB-β protein can inhibit the nuclear import of NF-κB/Rel proteins, but does not remove NF-κB/Rel proteins from the nucleus[17,18].
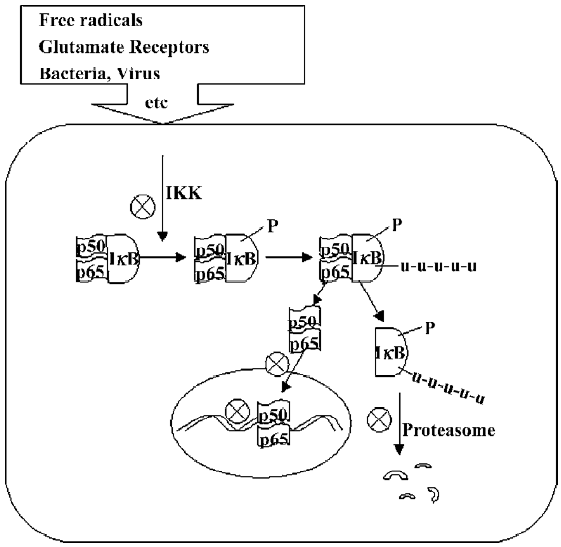
NF-κB activation pathway Signals that induce NF-κB activity cause the phosphorylation of IκB thereby activating the NF-κB complex. The activated NF-κB complex translocates into the nucleus and binds to DNA at the κB binding motifs and alters gene expression. Most signals that lead to the activation of NF-κB act on a high molecular weight complex containing a serine-specific IκB kinase (IKK). IKK contains at least 3 distinct subunits: IKK-α, IKK-β, and IKK-γ. IKK-α and IKK-β are catalytic kinase subunits, while IKK-γ is a regulator for sensing and integrating upstream activating signals[18]. There are 2 NF-κB activation pathways: the classical or canonical pathway and the non-canonical pathway. In the canonical pathway, the activation of the IKK complex leads to the phosphorylation of 2 specific serines (Ser32 and Ser36) in IκB-α, which targets IκB-α for ubiquitination and degradation by the 26S proteasome. In the non-canonical pathway, the p100–RelB complex is activated through phosphorylation by an IKK-α homodimer (lacking IKK-γ) to generate p52–RelB. In either pathway, the unmasked NF-κB complex can then enter the nucleus to activate target gene expression. In the classical pathway, one of the target genes activated by NF-κB can encode IκB-α, and newly-synthesized IκB-α enters the nucleus recombined with NF-κB, which can remove NF-κB from DNA, and export the complex back to the cytoplasm to restore the original latent state. Thus, the NF-κB activating pathway is a transient process, generally lasting from 30 to 60 min in most cells[20,21].
Dual roles of NF-κB in cell death and survival
NF-κB targets many genes to activate their expression. These target genes include cytokines/chemokines and their modulators, immunoreceptors, proteins involved in antigen presentation, cell adhesion molecules, acute-phase proteins, early response genes, stress response genes, cell surface receptors, transcription factors and regulators, regulators of apoptosis, growth factors, and cell death receptor ligands and their modulators. In the central nervous system (CNS), NF-κB can play an anti-apoptotic or pro-apoptotic role in cell death[22]. This is not unexpected as NF-κB regulates the genes involved in neuronal death and survival.
Anti-apoptotic activity of NF-κB A large number of studies have demonstrated that NF-κB plays a prosurvival role in proliferating cells, including tumor cells. Two actions of NF-κB make it an important cell survival transcription factor in these cells: the regulation of the cell cycle and the inhibition of apoptosis. The best example for elucidating the prosurvival action of NF-κB in cells is finding an inhibitory role of NF-κB in death receptor-induced apoptosis[23]. Binding to death receptors by TNF-α activates caspase-8 through TRAF1 and NF-κB through TRAF2. The activation of caspase-8 leads to apoptosis. Blocking NF-κB activation potentiates TNF-α-induced apoptosis, indicating NF-κB exerts anti-apoptotic action. It has been found that nerve growth factor (NGF) promotes neuronal survival through activating NF-κB[24]. Yu et al[25] reported that mice lacking the p50 subunit of NF-κB exhibited increased damage to hippocampal pyramidal neurons after the administration of the excitotoxin kainate. In immortalized mouse hippocampal cell line HT22 cells, glutamate-induced apoptosis was inhibited by IκB inhibitor aspirin, while the NF-κB decoy oligonucleotide potentiated it[26]. The action of NF-κB on neuronal survival is mediated through the upregulation of several prosurvival genes.
Superoxide dismutase Manganese superoxide dis-mutase (Mn-SOD) is an important antioxidant enzyme, which is a potent scavenger of superoxide anion and is likely to serve important cytoprotective roles against cellular damage. It has been reported that NF-κB is involved in the expression of Mn-SOD[27]. The incubation of human endometrial stromal cells with TNF-α or the phorbol 12-myristate 13-acetate (TPA), a protein kinase C activator, caused marked increases in nuclear NF-κB DNA binding activity and Mn-SOD mRNA and activity. These effects of TNF-α and TPA were completely inhibited by the proteasome inhibitor MG132 and the recombinant peptide capable of blocking NF-κB nuclear translocation, SN50[28]. Activities of Mn-SOD and SOD1 increased after spinal cord injury (SCI) and exposure to neurotoxins[29,30]. The increase in SOD appeared to be NF-κB-dependent, and overexpression significantly protected against the deleterious effect of reactive oxygen species, ceramide, or N-methyl-D-aspartate (NMDA). Several other studies have shown that the overexpression of copper-Zn SOD or the activation of Mn-SOD is neuropro-tective against ischemia, excitotoxicity, and Aβ toxicity[31–33].
Bcl-2 Bcl-2 and Bcl-XL are well-defined anti-apoptotic proteins. The NF-κB binding site is identified in the promoter of murine Bcl-x[34]. Bui et al[35] found that NGF increased the expression of Bcl-XL, possibly through the activation of NF-κB. Some studies indicate that TNF-α has neuroprotective effects. TNF-α increases the mRNA and protein levels of Bcl-2 and Bcl-x[36]. It has also been found that the exposure of cultured neurons to hypoxia/reoxygena-tion increases the levels of Bcl-2 and Bcl-X. The inhibition of NF-κB activation abolished the hypoxia-induced induction of Bcl-2 and Bcl-X, indicating that the induction of Bcl-2 and Bcl-X is mediated by NF-κB[37]. NF-κB is also reportedly involved in the activation of the Bcl-2 family member A1/Bfl-1[38].
Pro-apoptotic activity of NF-κB A large number of stu-dies have found that NF-κB activation participates in neuronal apoptosis. The mechanism by which NF-κB translocation induces apoptosis is not completely clear, but it is assumed that this mechanism involves the regulation of 1 or more genes known to play a pro-apoptotic role in apoptosis. Among the NF-κB-responsive genes possibly involved in the control of neuronal cell death, pro-apoptotic genes p53, c-Myc, cyclin D1, Bcl-Xs, and the Fas ligand and its receptor are activated by various pathological stimuli.
p53 The p53 protein is a tumor suppressor and plays important roles in neuronal apoptosis via promoting the expression of the pro-apoptotic gene Bax and PUMA, but suppresses the expression of the cytoprotective gene Bcl-2. NF-κB may contribute to neuronal apoptosis through the induction of p53. In the study of glutamate receptor-mediated excitotoxicity, upon stimulation of glutamate receptors, a quick and robust induction in the levels of p53 mRNA and protein was observed. The induction of p53 was blocked by NF-κB inhibitors[39,40]. Qin et al[40] investigated the role of NF-κB in apoptosis induced by the NMDA receptor agonists in rat striatal medium spiny neurons. The administration of the excitotoxin quinolic acid and NMDA induced apoptosis in the rat striatum. The inhibition of NF-κB nuclear translocation by the SN50, a recombinant cell permeable peptide containing the p50 nuclear localization sequence, reduced apoptotic death of striatal neurons and p53 expression. Uberti et al[41] pretreated the neuronal cultures with aspirin, which inhibits NF-κB activation, or with a specific p53 antisense oligonucleotide, which inhibits p53 transcription, resulting in a complete prevention of glutamate-induced p53 induction and apoptosis. The NF-κB-dependent induction of p53 was also found in response to DNA damage and oxidative stress. The induced p53 was apparently involved in cell death under these conditions as the synthetic p53 inhibitor pifithrin-α blocked neuronal apoptosis[42–45]. NF-κB not only regulates the levels of p53, but also increases the stability of the DNA binding of p53, providing an additional mechanism for promoting p53-mediated pro-apoptotic signaling[46].
Cyclin D1 and c-Myc The cyclins are a family of proteins that are involved in cell cycle progression and apoptosis. The best explored link between NF-κB activation and cell cycle progression involves cyclin D1, a cyclin which is expressed relatively early in the cell cycle and is crucial to DNA synthesis[47]. The NF-κB regulation of cyclin D1 occurs at the transcriptional level and is mediated by the direct binding of NF-κB to multiple sites in the cyclin D1 promoter[48]. NF-κB promotes G1 to S phase transition in mouse embryonic fibroblasts and in T47D mammary carcinoma cells[49]. The NF-κB-mediated induction of cyclin D1 was found in dorsal root ganglion neurons in response to ceramide-induced apoptosis. The inhibition of NF-κB blocked cyclin D1 induction and increased the viability of neurons[50]. Liang et al[44] reported that overstimulation NMDA receptors with quinolinic acid induced a NF-κB-dependent elevation in cyclin D1 mRNA and protein levels. The incorporation of BrdU was observed in some neurons undergoing apoptosis. NF-κB binding sites have also been identified in the c-Myc exon and upstream sites and positively regulate the expression of c-Myc[51]. In excitotoxic models, c-Myc was upregulated through NF-κB activation[39,44]. The NF-κB-dependent increase in c-Myc expression was also observed in 6-hydroxydopamine-induced Parkinson’s disease[45,52].
The involvement of cell cycle regulators in neuronal apoptosis has been shown by many investigators. The expression of certain cell cycle regulators, such as cyclin D1, cyclin G, c-Myc, and cdk4 have been found during neuronal apoptosis[53–56]. To support the role of cell cycle regulators in neuronal apoptosis, some studies showed that cdk inhibitors blocked neurotrophic factor withdrawal-induced apoptosis[57]. Cyclin D1 antisense or cell cycle inhibitors and cdk inhibitors partially blocked the excitotoxin-induced apoptosis of striatal neurons[44,58], suggesting cycle regulators play an important role in neuronal apoptosis.
Bcl-Xs and BAX The bcl-x gene functions to regulate cell death. Bcl-x transcripts are alternatively spliced into a long and short form or the form lacking the transmembrane domain. The long form (bcl-xL) represses cell death, while the short form (bcl-xs) favors apoptosis. NF-κB binding sites have been identified in the Bcl-x promoter[59]. The NF-κB-dependent induction in Bcl-Xs has been reported. Dixon et al[60] showed that following ischemia and NF-κB activation, Bcl-xs messenger RNA levels increase in the CA1 hippocampal region. In cultured endothelial cells, hypoxia decreased Bcl-2 mRNA levels, whereas the transfection of the NF-κB decoy significantly attenuated a decrease in Bcl-2 mRNA, increased Bcl-2/BAX ratio, and inhibited hypoxia-induced cell death[61]. The prolonged activation of NMDA receptors results in NF-κB nuclear translocation, release of LDH, increases in the BAX/Bcl-XL ratio, and DNA fragmentation. SN50 blocked the NMDA-induced increase in the Bax/Bcl-XL ratio and cell death[62]. Glutamate also reportedly increased the expression of BAX, which was inhabitable with BAY 11-7082, a selective inhibitor of IκB-α phosphorylation[63]. In cyanide-induced apoptosis, the expression levels of 2 anti-apoptotic Bcl-2 proteins, Bcl-2 and Bcl-XL, remained unchanged after cyanide treatment, whereas the mRNA levels of Bcl-Xs and Bax began to increase within 2 h, and their protein levels increased 6 h after treatment. Both NF-κB SN50 and the NF-κB decoy blocked the upregulation of Bcl-Xs and BAX[64]. In low potassium-induced apoptosis of cortical neurons, NF-κB DNA binding increased, and this was accompanied by an elevation in Bcl-Xs transcription. The latter was abolished by the inhibition of NF-κB or the restoration of potassium levels[65].
Nitric oxide The role of nitric oxide (NO) in apoptosis is complex, as it may exert proapoptotic or antiapoptotic effects depending on experimental conditions. NF-κB plays a role in regulating the expression of NO synthase (NOS). Xie et al[66] defined a NF-κB binding domain in murine inducible NOS (iNOS). NF-κB stimulated the expression of iNOS. The NF-κB inhibitor pyrrolidine dithiolidin inhibited the activation of NF-κB and the production of NO in lipopolysaccharide (LPS)-treated macrophages, suggesting that the activation of NF-κB/Rel is critical in the induction of iNOS by LPS.
NOS has been demonstrated to play a proapoptotic role in several in vitro and in vivo studies. The incubation of human breast cancer cell line MCF-7 cells and differentiated neuronal PC12 cells with TNF-α increased the expression and activity of iNOS. In addition to NOS inhibitors, iNOS antisense oligonucleotides effectively prevented NO2 generation and apoptosis, suggesting that the TNF-α-induced cell death is mediated by iNOS-derived NO[67,68]. Employing the intrastriatal injection of autologous blood in rats to model intracerebral hemorrhage, Zhao et al[69] demonstrated a robust and prolonged NF-κB activation and a robust induction of iNOS at both the mRNA and protein levels. In SCI models, iNOS was also found to be increased[70], and the drugs inhibiting NOS offered protective effects[71–73].
NF-κB inhibitors and neuroprotective therapy
Many human nervous system diseases have an association with NF-κB activation. These conditions, including aging[74], headache[75], pain[76], stroke[77], traumatic brain injury[78], SCI[79], Parkinson’s disease[80,81], multiple sclerosis[82], Alzheimer’s disease[83,84], amyotrophic lateral sclerosis[85], Huntington’s disease[86], and brain tumors[87–91], have been associated with the NF-κB pathway. As NF-κB plays important roles in regulating cell survival and death in a broad array of physiological and pathological conditions, it is an attractive proposal to manipulate NF-κB functions to obtain its beneficial effects or abolish its harmful actions when it is required[92]. Multiple signaling events are involved in NF-κB activation, including the phosphorylation and degradation of IκB, NF-κB nuclear translocation, and DNA interaction, thus making it a relatively easy target for drug actions (Figure 1). There are a large number of com-pounds have been reported to inhibit NF-κB functions. These compounds mainly include antioxidants, non-steroidal anti-inflammatory drugs (NSAID), flavonoids, protease inhibitors. A few of these compounds have been used in the clinical setting.
Antioxidants Oxidative stress is one of the common pathogenic mechanisms in neurodegenerative disorders. Thus, antioxidants are frequently employed in the treatment of several neurodegenerative diseases, and are the most valuable therapeutic strategy for fighting neurodegeneration. Although it is hard to attribute a single mechanism to any antioxidants’ neuroprotective effects, the inhibition of NF-κB activation is a prominent feature of antioxidants. The antioxidants include N-acetyl-L-cysteine (NAC), α-lipoic acid, glutathione monoester, pyrrolidine dithiocarbamate (PDTC), tepoxalin, and flavonoids.
Free radicals are important mediators for NF-κB activation. NAC, a well-characterized antioxidant, is found to exert neuro-protective effects against free radical-related neuronal injury[93,94]. NAC influences many cellular signaling pathways, including c-Jun N-terminal kinase, p38 mitogen-activated protein kinase, and redox-sensitive activating protein-1. NAC can also prevent apoptosis and promote cell survival by activating the extracellular signal-regulated kinase pathway. NAC directly modifies the activity of several proteins by its reducing activity[95], and is demonstrated to inhibit the degradation of IκB-α and the activation of NF-κB[96–98]. In animal models of global ischemia, pretreatment with NAC (300 mg/kg) or another antioxidant PDTC (200 mg/kg) significantly reduced the infarct volume. NAC has also been reported to increase the survival of dopaminergic neurons. The local or systemic administration of NAC protected dopamine neurons against 6-hydroxydopamine-induced oxidative damage[99].PDTC is an antioxidant that has been studied for many years. Using cell cultures, Schreck et al[100] found that micromolar concentrations of PDTC reversibly suppressed NF-κB activation. PDTC specifically prevented the NF-κB-dependent transactivation of reporter genes under the control of the HIV-1 long terminal repeat and simian virus 40 enhancer. In other studies, PDTC inhibited NF-κB activation while enhancing the binding activity of activator protein-1[101]. In addition, PDTC can inhibit the NF-κB-mediated production of TNF-α, hypoxia-induced dephosphorylation of Akt, and inflammatory responses[102–104]. It has been shown that treatment with PDTC significantly attenuates reperfusion-induced lung injury[105], glycerol-induced renal injury[106], cholestatic liver injury[107], and adriamycin-induced myocardial apoptosis[108]. Crack et al[109] observed that knockout glutathione peroxidase-1 (Gpx1) in mice increased the ischemia-induced activation of NF-κB. PDTC was able to afford partial neuroprotection in the Gpx1-null mice. In Wistar rats, PDTC prevented NF-κB activation in the ischemic brain, as determined by the reduced DNA binding and nuclear translocation of NF-κB in neurons. PDTC treatment reduced the infarction volume by 48% when given 6 h after MCAO[110].
Flavonoids are potent antioxidants found in many natural products. They are widely used as food supplements and as anti-inflammation and antitumor drugs. Recently, many studies have demonstrated that flavonoids have neuroprotective effects in animal models[46]. Among them, Ginkgo biloba has received particular attention. Chen et al[111] found that Ginkgo biloba extract significantly reduced intracellular reactive oxygen species formation and NF-κB activation induced by TNF-α. Tea extracts have been previously reported to possess radical scavenger, iron chelating, and anti-inflammatory properties in a variety of tissues. Recent studies found that green tea extracts were capable of inhibiting NF-κB[112]. Studies demonstrated that green tea extracts inhibited iron-induced lipid peroxidation, NF-κB activation, and 6-hydroxydopamine (6-OHDA)-induced neuronal death. 6-OHDA-induced apoptosis of catecholaminergic PC12 cells was inhibited by green tea polyphenols and their major effective component epigallocatechin-3-gallate at a concentration of 200 mmol/L[113]. Green tea polyphenols have been shown to reduce the toxic effects of β-amyloid, ischemia/reperfusion-induced apoptosis, and the infarct volume[114,115]. Given by brain penetrating property of polyphenols, these compounds may be utilized as a class of drugs for the treatment of neurodegenerative diseases.
IκB phosphorylation and degradation inhibitors Phosphorylation and the subsequent proteasomal degradation of IκB are key steps for NF-κB activation. NSAID and cyclopentone prostaglandins are now found to be IκB inhibitors. In 1994, Kopp and Ghosh[116] reported that sodium salicylate and aspirin inhibited the activation of NF-κB through blocking the degradation of the IκB. IKK-α and IKK-β phosphorylate IκB. Aspirin and sodium salicylate can inhibit IKK-β activity in vitro and in vivo. The mechanism by which aspirin and sodium salicylate inhibit NF-κB is the binding of these agents to IKK-β to reduce ATP binding[117]. These studies not only further explain the new mechanism of actions, but also suggest new implications of these drugs. Grilli et al[118] found that acetylsalicylic acid and its metabolite sodium salicylate protected neurons against neurotoxicity elicited by the excitatory amino acid glutamate in rat primary neuronal cultures and hippocampal slices. This inhibitory effect may be involved in the inhibition of NF-κB activation, protein kinase C zeta activity, superoxide anion generation, and lipid peroxidation[118–120]. Recent studies suggest that aspirin and other NSAID protected cultured mesencephalic cells against 6-OHDA, 1-methyl-4 phenylpyri-dinium, and glutamate-induced toxicity[121,122]. Using NSAID in Parkinson’s disease (PD) has also been proposed[123]. It has been found that arthritis patients taking aspirin have a lower incident and later onset of Alzheimer’s disease (AD). Emerging evidence shows that aspirin and other NSAID have multiple influences on the AD pathogenic process, including inhibiting the formation of fibrillar Aβ, destabilizing preformed fibrillar β-amyloid (Aβ), and preventing the aggregation of Aβ and attenuating its toxicity[124–126]. Recent studies also indicate that acetylsalicylic acid inhibits tau phosphorylation[127]. Other potentially interesting anti-inflammatory drugs have also been reported to exert neuroprotective effects and inhibit NF-κB, including cannabinoid dexanabinol and caffeic acid[128–130].
Recent studies have identified that cyclopentenone prostaglandins (cPG), including prostaglandin (PG)A1 and PGJ2 are ligands for peroxisome-proliferation activator receptor-γ and inhibitors of NF-κB. Rossi et al[131] reported that PGA1 could block NF-κB activation by inhibiting IκB phos-phorylation. In a subsequent study, they further identified that PGA1 could directly inhibit IκB kinase-β[132]. PGA1 also increases the expression of IκB-α[133]. Similar inhibitory effects of PGJ2 and PGE1 on NF-κB were observed[134–136]. In addition, PGJ2 was found to interfere with DNA binding through covalently modifying the NF-κB p50 subunit[137]. Thus, cPG inhibit NF-κB by interfering in multiple sites in the NF-κB signaling pathway from IκB synthesis to DNA binding[138]. Studies have shown that cPG have neuroprotective effects under certain pathological conditions. Qin et al[139] first reported that PGA1 protected striatal neurons against NMDA receptor agonist quinolinic acid-induced apoptosis through inhibiting NF-κB activation. PGA1 also inhibited the mitochondrial toxin rotenone-induced death of dopaminergic cell line SH-SY5Y cells[140]. In recent studies, PGA1 and PGJ2 were reported to reduce ischemic brain damage through inducing heat shock proteins, inhibiting NF-κB activation, and inflammation[141–143]. These studies revealed that the administration of PGA1 and PGJ2 2–3 h post-ischemia was still effective. Other studies have also reported that PGJ2 reduced ischemic myocardial infarction[144].
Other drugs, such as estrogen, curcumin, and quercetin have been reported to inhibit IκB degradation[145–148]. All these compounds are reported to have neuroprotective effects under certain experimental conditions.
NF-κB nuclear translocation and DNA binding inhibitors NF-κB has to translocate into the nucleus in order to regulate gene expression. Drugs acting on NF-κB nuclear transport and DNA binding have received considerable attention. Lin et al[149] synthesized a membrane-permeable recombinant peptide NF-κB SN50. This peptide contains a signal peptide, which confers the cell membrane permeability of SN50, and a nuclear localization signal, which competes with NF-κB for nuclear entry. The peptide inhibits the nuclear translocation of NF-κB in cultured endothelial and monocytic cells stimulated with LPS or TNF-α in a concentration-dependent manner. This peptide has been widely used for dissecting cellular functions of NF-κB[150,151]. We, along with others, have successfully used SN50 for inhibiting NF-κB nuclear import in vivo and have found that it is very effective in blocking NF-κB nuclear entry and NF-κB-mediated target gene transcription in response to various stimuli[39,44,45,150,152–156]. An intrastriatal or nigral injection of SN50 substantially inhibited the nuclear translocation of NF-κB, inhibiting the expression of NF-κB target genes p53, c-Myc and cyclin D1, and attenuating excitotoxicity[39,44,45,153]. One study has successfully blocked cholecystokinin-octapeptide-induced pancreatitis with an intraperitoneal injection of SN50[155]. These studies suggest that nuclear import inhibitors represent an important class of NF-κB inhibitors.
Many studies have focused on NF-κB DNA decoys. The NF-κB DNA decoys are double-stranded DNA oligonucleotides (ODN) containing the NF-κB-binding motif. When delivered to cells, it competes with NF-κB for DNA binding sites, and thus inhibits NF-κB function. Some studies have demonstrated that ODN that are delivered locally or systemically are effective in blocking NF-κB transactivation activity[157–160]. Some in vivo therapeutic studies with ODN have generated promising results. The systemic administration of ODN suppressed NF-κB activity, and the expression of cytokines protected liver grafts against ischemia/reperfusion-induced injury in rats[161]. The animal studies showed that NF-κB decoys reduced lung vascular permeability in septic mice, improved lung function[162], and inhibited hepatic metastasis in the mice loaded with murine reticulosarcoma M5076[163]. The clinical usefulness of ODN has been tested in 2 patients with percutaneous coronary intervention. The initial results showed the suppression of restenosis with no observed adverse effect[164]. NF-κB decoys may be a potential therapeutic strategy for certain types of diseases[165,166].
Drugs that directly inhibit NF-κB DNA binding have been found, but have not been well characterized. For example, it has been reported that the metal-chelating drug aurine tricarboxylic acid inhibited NF-κB DNA interaction[167].
Neurological disorders may benefit from NF-κB inhibitors
Ischemic brain injury The distribution of NF-κB was investigated immunohistochemically in post-mortem brains of stroke patients. An enhanced immunoreactivity of NF-κB was observed in glial cells of infarcted areas, particularly in the penumbra or border zone between the ischemic and non-ischemic areas[168]. In animal studies, early activation of NF-κB has been found to precede DNA damage after ischemic attack[169,170]. It was reported that ischemia induced a TNF-like weak inducer of apoptosis (TWEAK) and its membrane receptor Fn14. TWEAK promotes neuronal cell death and activates NF-κB through the upstream kinase IKK[171]. The deletion of the neuronal IKK2 subunit or inhibition of IKK activity reduced the infarct size and neuronal cell loss. The role of NF-κB in neuronal death was further suggested, as several neuroprotective agents, such as antioxidant LY231617, PDTC, and PGA1, have been shown to inhibit NF-κB activation, reduce infarct volume, and improve behavior deficits[110,173,174]. The contribution of NF-κB activation to ischemic neuronal damage has also been assessed with either the expression of mutant IκB-α in neurons and glial, or NF-κB p50 knockout mice and transgenic mice. The results indicated that the neuronal expression of the NF-κB inhibitor reduced both the infarct size and cell death[174]. Mice lacking the p50 subunit of NF-κB develop significantly smaller infarcts after transient focal ischemia[173,175]. These studies may have established the rationale for use of NF-κB inhibitors in ischemic brain injury.
PD Recently, the role of the neuron-glia interaction and the inflammatory process in PD has been the focus of intense study by the research community. The increase in NF-κB has been found in the post-mortem brains of PD patients. In PD patients, the proportion of nigral dopaminergic neurons with immunoreactive NF-κB and interferon-γ was significantly increased in comparison with control patients[81,176]. A possible relationship between the nuclear localization of NF-κB in the mesencephalic neurons of PD patients and oxidative stress in such neurons has been shown in vitro with primary cultures of rat mesencephalon, where the translocation of NF-κB is preceded by a transient production of free radicals during apoptosis induced by the activation of the sphingomyelin-dependent signaling pathway with C2-ceramide. The data suggest that this oxidant-mediated apoptogenic transduction pathway may play a role in the mechanism of neuronal death in PD[176]. In animal models of PD, the inhibition of NF-κB achieves neuroprotection against the 6-OHDA- and MPTP-induced degeneration of dopaminergic neurons[44,45,112,177], suggesting that NF-κB inhibitors could be beneficial in PD.
AD The distribution of NF-κB was investigated immuno-histochemically in the post-mortem cases of AD. In the AD cases, increased staining for NF-κB p65 was seen in neurons and their processes, neurofibrillary tangles, and dystrophic neurites. The neuronal staining observed in AD was strongest in the hippocampal formation and entorhinal cortex[178]. Boissière et al[179] studied the cellular distribution of NF-κB in the nucleus basalis of Meynert of AD and control patients. The proportion of large cholinergic neurons with elevated nuclear immunostaining of NF-κB was significantly increased in AD, suggesting an association between NF-κB functions and the process of cholinergic degeneration in AD. In another report, NF-κB immunoreactivity was found in the neutrophil of diffuse Aβ deposits. In addition, NF-κB immunoreactivity was found in the nuclei of neurons, but not in the nuclei of reactive astrocytes, in the vicinity of diffuse plaques[180]. The discovery of NF-κB activation in AD has been confirmed by other investigators[181–183]. Since inflammation is a prominent feature in AD, NF-κB may participate in the inflammatory response. A more direct connection of AD pathogenesis and NF-κB was observed as Aβ activates NF-κB[184–186]. Although an early in vitro study found that the activation of NF-κB by a low dose of Aβ may have neuroprotective effects[187], other studies have been observed that NF-κB inhibitors inhibit the production of Aβ[188]. In rat primary neurons and human post-mitotic neuronal cells, the Aβ peptide induced dose-dependent neuronal death, the nuclear translocation of the p65 and p50 subunits, and an apoptotic profile of gene expression. The anti-inflammatory drug aspirin and the selective IκB kinase 2 inhibitor AS602868 completely inhibited p50/p65 nuclear translocation and neuronal damage[189]. The clinical trials with some NSAID did not generate encouraging outcomes, as noted by Valerio et al[190], since not all NSAID can inhibit Aβ production. Better compounds with the ability to reduce Aβ should be selected in future studies.
Excitotoxicity Excitotoxicity has been implicated in several neurodegenerative diseases. Kaltschmidt et al[191] reported that the ionotropic glutamate receptor agonist kainic acid (KA) activates NF-κB. Later studies defined a pro-apoptotic role of NF-κB activation and nuclear translocation mediated by AMPA/KA receptors[153,192]. Similarly, the stimulation of glutamate NMDA receptors robustly activates NF-κB through the degradation of IκB-α[139,152]. In other studies, the pharmacological upregulation of NF-κB increased glutamate-induced excitotoxicity, while the upregulation of CREB decreased excitotoxicity[193]. Grilli et al[118] reported a neuroprotective role of aspirin on the glutamate-induced death of hippocampal neurons, opening a new avenue for the study of excitotoxicity. Since then, several studies have reported that the inhibition of NF-κB has neuroprotective effects. In studies conducted by Casper et al[121], neuroprotection against glutamate-mediated excitotoxicity was also found with ibuprofen. The inhibition of NF-κB with a herbal active component glycyrrhiza acid[193], free radical scavenger OCT14117[194], and glutamate metabotropic receptor agonists (2S, 1’S,2’S)-(carboxy-cyclopropyl)glycine and amino-4-phosphonobutyric acid[195] was associated with a neuroprotective effect. These results suggest that NF-κB inhibitors could be suitable drugs for blocking excitotoxicity.
Summary
The signaling pathway and the role of NF-κB have been studied for more than 2 decades. However, we still have limited knowledge on the role of NF-κB in CNS neurons and the molecular mechanisms underlying its actions. Many controversial findings need to be consolidated. In particular, its dual roles in neuronal death and survival and underlying molecular mechanisms need to be carefully evaluated in relation to human neurological diseases[197–200]. The involvement of NF-κB in human diseases certainly establishes it as a potential target for therapy. Many common synthetic (eg aspirin) and traditional remedies target, at least in part, the NF-κB signaling. Our knowledge of the molecular details of the NF-κB pathway will enable us to develop more specific NF-κB inhibitors to treat neurological diseases.
References
- Baldwin AS Jr. The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol 1996;14:649-83.
- Shimada M, Satoh N, Yokosawa H. Involvement of Rel/NF-kappaB in regulation of ascidian notochord formation. Dev Growth Differ 2001;43:145-54.
- Weih F, Caamaño J. Regulation of secondary lymphoid organ development by the nuclear factor-kappaB signal transduction pathway. Immunol Rev 2003;195:91-105.
- Sen R, Baltimore D. Inducibility of k immunoglobulin enhancer-binding protein NF-κB by a post-translational mechanism. Cell 1986;47:921-8.
- O’Neill LA, Kaltschmidt C. NF-kappa B: a crucial transcription factor for glial and neuronal cell function. Trends Neurosci 1997;20:252-8.
- O’Mahony A, Raber J, Montano M, Foehr E, Han V, Lu SM, et al. NF-kappaB/Rel regulates inhibitory and excitatory neuronal function and synaptic plasticity. Mol Cell Biol 2006;26:7283-98.
- Meffert MK, Chang JM, Wiltgen BJ, Fanselow MS, Baltimore D. NF-kappa B functions in synaptic signaling and behavior. Nat Neurosci 2003;6:1072-8.
- Mémet S. NF-kappaB functions in the nervous system: from development to disease. Biochem Pharmacol 2006;72:1180-95.
- Xiao G, Rabson AB, Young W, Qing G, Qu Z. Alternative pathways of NF-kappaB activation: a double-edged sword in health and disease. Cytokine Growth Factor Rev 2006;17:281-93.
- Denk A, Wirth T, Baumann B. NF-kappaB transcription factors: critical regulators of hematopoiesis and neuronal survival. Cytokine Growth Factor Rev 2000;11:303-20.
- Grilli M, Memo M. Nuclear factor-kappaB/Rel proteins: a point of convergence of signalling pathways relevant in neuronal function and dysfunction. Biochem Pharmacol 1999;57:1-7.
- Mattson MP, Meffert MK. Roles for NF-kappaB in nerve cell survival, plasticity, and disease. Cell Death Differ 2006;13:852-60.
- Cornwell WD, Kirkpatrick RB. Cactus-independent nuclear translocation of Drosophila RELISH. J Cell Biochem 2001;82:22-37.
- Lätzer J, Papoian GA, Prentiss MC, Komives EA, Wolynes PG. Induced fit, folding, and recognition of the NF-kappaB-nuclear localization signals by IkappaBalpha and IkappaBbeta. J Mol Biol 2007;367:262-74.
- Heissmeyer V, Krappmann D, Hatada EN, Scheidereit C. Shared pathways of IκBα kinase-induced SCF TrCP-mediated ubiquitina-tion and degradation for the NF-κB precursor p105 and IκBα. Mol Cell Biol 2001;21:1024-35.
- Dejardin E. The alternative NF-kappaB pathway from biochemistry to biology: pitfalls and promises for future drug development. Biochem Pharmacol 2006;72:1161-79.
- Baeuerle PA. I-kappa-B — NF-kappa-B structures: at the interface of inflammation control. Cell 1998;95:729-31.
- Lee SH, Hannink M. Characterization of the nuclear import and export functions of IkappaBalpha. J Biol Chem 2002;26:23358-66.
- Piette J, Piret B, Bonizzi G, Schoonbroodt S, Merville MP, Legrand-Poels S, et al. Multiple redox regulation in NF-kappaB transcription factor activation. Biol Chem 1997;378:1237-45.
- Huynh QK, Kishore N, Mathialagan S. Kinetic mechanisms of IkappaB-related kinases (IKK) inducible IKK and TBK-1 differ from IKK-1/IKK-2 heterodimer. J Biol Chem 2002;277:12550-8.
- Gilmore TD. Introduction to NF-κB: players, pathways, perspectives. Oncogene 2006;25:6680-4.
- Kaltschmidt B, Widera D, Kaltschmidt C. Signaling via NF-kappaB in the nervous system. Biochim Biophys Acta 2005; 1745: 287−99.
- Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS Jr. NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science 1998;281:1680-3.
- Maggirwar SB, Sarmiere PD, Dewhurst S, Freeman RS. Nerve growth factor-dependent activation of NF-kappaB contributes to survival of sympathetic neurons. J Neurosci 1998;18:10356-65.
- Yu Z, Zhou D, Bruce-Keller AJ, Kindy MS, Mattson MP. Lack of the p50 subunit of nuclear factor-kappaB increases the vulnerability of hippocampal neurons to excitotoxic injury. J Neurosci 1999;19:8856-65.
- Ishige K, Tanaka M, Arakawa M, Saito H, Ito Y. Distinct nuclear factor-kappaB/Rel proteins have opposing modulatory effects in glutamate-induced cell death in HT22 cells. Neurochem Int 2005;47:545-55.
- Maehara K, Hasegawa T, Isobe KI. A NF-kappaB p65 subunit is indispensable for activating manganese superoxide: dismutase gene transcription mediated by tumor necrosis factor-alpha. J Cell Biochem 2000;77:474-86.
- Sugino N, Karube-Harada A, Sakata A, Takiguchi S, Kato H. Nuclear factor-kappa B is required for tumor necrosis factor-alpha-induced manganese superoxide dismutase expression in human endometrial stromal cells. J Clin Endocrinol Metab 2002;87:3845-50.
- Borg J, London J. Copper/zinc superoxide dismutase overexpres-sion promotes survival of cortical neurons exposed to neurotoxins in vitro. J Neurosci Res 2002;70:180-9.
- Yune TY, Lee SM, Kim SJ, Park HK, Oh YJ, Kim YC, et al. Manganese superoxide dismutase induced by TNF-beta is regulated transcriptionally by NF-kappaB after spinal cord injury in rats. J Neurotrauma 2004;21:1778-94.
- Mattson MP, Goodman Y, Luo H, Fu W, Furukawa K. Activation of NF-kappaB protects hippocampal neurons against oxidative stress-induced apoptosis: evidence for induction of manganese superoxide dismutase and suppression of peroxynitrite production and protein tyrosine nitration. J Neurosci Res 1997;49:681-97.
- Saito A, Hayashi T, Okuno S, Ferrand-Drake M, Chan PH. Overex-pression of copper/zinc superoxide dismutase in transgenic mice protects against neuronal cell death after transient focal ischemia by blocking activation of the Bad cell death signaling pathway. J Neurosci 2003;23:1710-8.
- Sompol P, Xu Y, Ittarat W, Daosukho C, St Clair D. NF-kappaB-associated MnSOD induction protects against beta-amyloid-induced neuronal apoptosis. J Mol Neurosci 2006;29:279-88.
- Glasgow JN, Wood T, Perez-Polo JR. Identification and characterization of nuclear factor kappaB binding sites in the murine bcl-x promoter. J Neurochem 2000;75:1377-89.
- Bui NT, Livolsi A, Peyron JF, Prehn JH. Activation of nuclear factor kappaB and Bcl-x survival gene expression by nerve growth factor requires tyrosine phosphorylation of IkappaBalpha. J Cell Biol 2001;152:753-64.
- Tamatani M, Che YH, Matsuzaki H, Ogawa S, Okado H, Miyake S, et al. Tumor necrosis factor induces Bcl-2 and Bcl-x expression through NFkappaB activation in primary hippocampal neurons. J Biol Chem 1999;274:8531-8.
- Tamatani M, Mitsuda N, Matsuzaki H, Okado H, Miyake S, Vitek MP, et al. A pathway of neuronal apoptosis induced by hypoxia/reoxygenation: roles of nuclear factor-kappaB and Bcl-2. J Neurochem 2000;75:683-93.
- Wang CY, Guttridge DC, Mayo MW, Baldwin AS Jr. NF-kappaB induces expression of the Bcl-2 homologue A1/Bfl-1 to preferentially suppress chemotherapy-induced apoptosis. Mol Cell Biol 1999;19:5923-9.
- Qin ZH, Chen RW, Wang Y, Nakai M, Chuang DM, Chase TN. Nuclear factor kappaB nuclear translocation upregulates c-Myc and p53 expression during NMDA receptor-mediated apoptosis in rat striatum. J Neurosci 1999;19:4023-33.
- Grilli M, Memo M. Possible role of NF-kappaB and p53 in the glutamate-induced pro-apoptotic neuronal pathway. Cell Death Differ 1999;6:22-7.
- Uberti D, Grilli M, Memo M. Contribution of NF-kappaB and p53 in the glutamate-induced apoptosis. Int J Dev Neurosci 2000;18:447-54.
- Culmsee C, Zhu X, Yu QS, Chan SL, Camandola S, Guo Z, et al. A synthetic inhibitor of p53 protects neurons against death induced by ischemic and excitotoxic insults, and amyloid beta-peptide. J Neurochem 2001;77:220-8.
- Aleyasin H, Cregan SP, Iyirhiaro G, O’Hare MJ, Callaghan SM, Slack RS, et al. Nuclear factor-(kappa)B modulates the p53 response in neurons exposed to DNA damage. J Neurosci 2004;24:2963-73.
- Liang ZQ, Wang X, Li LY, Wang Y, Chen RW, Chuang DM, et al. Nuclear factor-kappaB-dependent cyclin D1 induction and DNA replication associated with N-methyl-D-aspartate receptor-mediated apoptosis in rat striatum. J Neurosci Res 2007;85:1295-309.
- Liang ZQ, Li YL, Zhao XL, Han R, Wang XX, Wang Y, et al. NF-kappaB contributes to 6-hydroxydopamine-induced apoptosis of nigral dopaminergic neurons through p53. Brain Res 2007;1145:190-203.
- Fujioka S, Schmidt C, Sclabas GM, Li Z, Pelicano H, Peng B, et al. Stabilization of p53 is a novel mechanism for proapoptotic function of NF-kappaB. J Biol Chem 2004;279:27549-59.
- Joyce D, Albanese C, Steer J, Fu M, Bouzahzah B, Pestell RG. NF-kappaB and cell-cycle regulation: the cyclin connection. Cytokine Growth Factor Rev 2001;12:73-90.
- Guttridge DC, Albanese C, Reuther JY, Pestell RG, Baldwin AS Jr. NF-kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol Cell Biol 1999;19:5785-99.
- Hinz M, Krappmann D, Eichten A, Heder A, Scheidereit C, Strauss M. NF-kappaB function in growth control: regulation of cyclin D1 expression and G0/G1-to-S-phase transition. Mol Cell Biol 1999;19:2690-8.
- Gill JS, Windebank AJ. Ceramide initiates NFkappaB-mediated caspase activation in neuronal apoptosis. Neurobiol Dis 2000;7:448-61.
- Ji L, Arcinas M, Boxer LM. NF-kappa B sites function as positive regulators of expression of the translocated c-myc allele in Burkitt’s lymphoma. Mol Cell Biol 1994;14:7967-74.
- Tarabin V, Schwaninger M. The role of NF-kappaB in 6-hydroxydopamine- and TNFalpha-induced apoptosis of PC12 cells. Naunyn Schmiedebergs Arch Pharmacol 2004;369:563-9.
- Timsit S, Rivera S, Ouaghi P, Guischard F, Tremblay E, Ben-Ari Y, et al. Increased cyclin D1 in vulnerable neurons in the hippocampus after ischaemia and epilepsy: a modulator of in vivo programmed cell death? Eur J Neurosci 1999;11:263-78.
- Di Giovanni S, Knoblach SM, Brandoli C, Aden SA, Hoffman EP, Faden AI. Gene profiling in spinal cord injury shows role of cell cycle in neuronal death. Ann Neurol 2003;53:454-68.
- Wen Y, Yang S, Liu R, Simpkins JW. Cell-cycle regulators are involved in transient cerebral ischemia induced neuronal apoptosis in female rats. FEBS Lett 2005;579:4591-9.
- Rao HV, Thirumangalakudi L, Desmond P, Grammas P. Cyclin D1, cdk4, and Bim are involved in thrombin-induced apoptosis in cultured cortical neurons. J Neurochem 2007;101:498-505.
- Appert-Collin A, Hugel B, Levy R, Niederhoffer N, Coupin G, Lombard Y, et al. Cyclin dependent kinase inhibitors prevent apoptosis of postmitotic mouse motoneurons. Life Sci 2006;79:484-90.
- Ino H, Chiba T. Cyclin-dependent kinase 4 and cyclin D1 are required for excitotoxin-induced neuronal cell death in vivo. J Neurosci 2001;21:6086-94.
- Glasgow JN, Qiu J, Rassin D, Grafe M, Wood T, Perez-Pol JR. Transcriptional regulation of the BCL-X gene by NF-kappaB is an element of hypoxic responses in the rat brain. Neurochem Res 2001;26:647-59.
- Dixon EP, Stephenson DT, Clemens JA, Little SP. Bcl-Xshort is elevated following severe global ischemia in rat brains. Brain Res 1997;776:222-9.
- Matsushita H, Morishita R, Nata T, Aoki M, Nakagami H, Taniyama Y, et al. Hypoxia-induced endothelial apoptosis through nuclear factor-kappaB (NF-kappaB)-mediated bcl-2 suppression: in vivo evidence of the importance of NF-kappaB in endothelial cell regulation. Circ Res 2000;86:974-81.
- McInnis J, Wang C, Anastasio N, Hultman M, Ye Y, Salvemini D, et al. The role of superoxide and nuclear factor-kappaB signaling in N-methyl-D-aspartate-induced necrosis and apoptosis. J Pharmacol Exp Ther 2002;301:478-87.
- Pizzi M, Sarnico I, Boroni F, Benetti A, Benarese M, Spano PF. Inhibition of IkappaBalpha phosphorylation prevents glutamate-induced NF-kappaB activation and neuronal cell death. Acta Neurochir Suppl 2005;93:59-63.
- Shou Y, Li N, Li L, Borowitz JL, Isom GE. NF-kappaB-mediated up-regulation of Bcl-X(S) and Bax contributes to cytochrome c release in cyanide-induced apoptosis. J Neurochem 2002;81:842-52.
- Tao Y, Yan D, Yang Q, Zeng R, Wang Y. Low K+ promotes NF-kappaB/DNA binding in neuronal apoptosis induced by K+ loss. Mol Cell Biol 2006;26:1038-50.
- Xie QW, Kashiwabara Y, Nathan C. Role of transcription factor NF-kappa B/Rel in induction of nitric oxide synthase. J Biol Chem 1994;269:4705-8.
- Heneka MT, Löschmann PA, Gleichmann M, Weller M, Schulz JB, Wüllner U, et al. Induction of nitric oxide synthase and nitric oxide-mediated apoptosis in neuronal PC12 cells after stimulation with tumor necrosis factor-alpha/lipopolysaccharide. J Neurochem 1998;71:88-94.
- Binder C, Schulz M, Hiddemann W, Oellerich M. Induction of inducible nitric oxide synthase is an essential part of tumor necrosis factor-alpha-induced apoptosis in MCF-7 and other epithelial tumor cells. Lab Invest 1999;79:1703-12.
- Zhao X, Zhang Y, Strong R, Zhang J, Grotta JC, Aronowski J. Distinct patterns of intracerebral hemorrhage-induced alterations in NF-kappaB subunit, iNOS, and COX-2 expression. J Neurochem 2007;101:652-63.
- Miscusi M, Ebner F, Ceccariglia S, Menegazzi M, Mariotto S, Berra L, et al. Early nuclear factor-kappaB activation and inducible nitric oxide synthase expression in injured spinal cord neurons correlating with a diffuse reduction of constitutive nitric oxide synthase activity. J Neurosurg Spine 2006;4:485-93.
- Hecker M, Preiss C, Klemm P, Busse R. Inhibition by antioxidants of nitric oxide synthase expression in murine macrophages: role of nuclear factor kappa B and interferon regulatory factor 1. Br J Pharmacol 1996;118:2178-84.
- Liu X, Buffington JA, Tjalkens RB. NF-kappaB-dependent production of nitric oxide by astrocytes mediates apoptosis in differentiated PC12 neurons following exposure to manganese and cytokines. Brain Res Mol Brain Res 2005;141:39-47.
- Pokharel YR, Liu QH, Oh JW, Woo ER, Kang KW. 4-Hydroxykobusin inhibits the induction of nitric oxide synthase by inhibiting NF-kappaB and AP-1 activation. Biol Pharm Bull 2007;30:1097-101.
- Chung HY, Kim HJ, Kim KW, Choi JS, Yu BP. Molecular inflammation hypothesis of aging based on the anti-aging mechanism of calorie restriction. Microsc Res Tech 2002;59:264-72.
- Reuter U, Chiarugi A, Bolay H, Moskowitz MA. Nuclear factor-kappaB as a molecular target for migraine therapy. Ann Neurol 2002;51:507-16.
- Tegeder I, Niederberger E, Schmidt R, Kunz S, Gühring H, Ritzeler O, et al. Specific Inhibition of IkappaB kinase reduces hyperalgesia in inflammatory and neuropathic pain models in rats. J Neurosci 2004;24:1637-45.
- Herrmann O, Baumann B, de Lorenzi R, Muhammad S, Zhang W, Kleesiek J, et al. IKK mediates ischemia-induced neuronal death. Nat Med 2005;11:1322-9.
- Hang CH, Shi JX, Li JS, Wu W, Yin HX. Concomitant upregulation of nuclear factor-kappaB activity, proinflammatory cytokines and ICAM-1 in the injured brain after cortical contusion trauma in a rat model. Neurol India 2005;53:312-7.
- Brambilla R, Bracchi-Ricard V, Hu WH, Frydel B, Bramwell A, Karmally S, et al. Inhibition of astroglial nuclear factor kappaB reduces inflammation and improves functional recovery after spinal cord injury. J Exp Med 2005;202:145-56.
- Soós J, Engelhardt JI, Siklós L, Havas L, Majtényi K. The expression of PARP, NF-kappa B and parvalbumin is increased in Parkinson disease. Neuroreport 2004;15:1715-8.
- Mogi M, Kondo T, Mizuno Y, Nagatsu T. p53 protein, interferon-gamma, and NF-kappaB levels are elevated in the parkinsonian brain. Neurosci Lett 2007;414:94-7.
- Satoh J, Illes Z, Peterfalvi A, Tabunoki H, Rozsa C, Yamamura T. Aberrant transcriptional regulatory network in T cells of multiple sclerosis. Neurosci Lett 2007;422:30-3.
- Mattson MP, Camandola S. NF-kappaB in neuronal plasticity and neurodegenerative disorders. J Clin Invest 2001;107:247-54.
- Collister KA, Albensi BC. Potential therapeutic targets in the NF-kappaB pathway for Alzheimer’s disease. Drug News Perspect 2005;18:623-9.
- Xu Z, Chen S, Li X, Luo G, Li L, Le W. Neuroprotective effects of (-)-epigallocatechin-3-gallate in a transgenic mouse model of amyotrophic lateral sclerosis. Neurochem Res 2006;31:1263-9.
- Khoshnan A, Ko J, Watkin EE, Paige LA, Reinhart PH, Patterson PH. Activation of the IkappaB kinase complex and nuclear factor-kappaB contributes to mutant huntingtin neurotoxicity. J Neurosci 2004;24:7999-8008.
- Hayashi S, Yamamoto M, Ueno Y, Ikeda K, Ohshima K, Soma G, et al. Expression of nuclear factor-kappa B, tumor necrosis factor receptor type 1, and c-Myc in human astrocytomas. Neurol Med Chir (Tokyo) 2001;41:187-95.
- Bian X, Opipari AW Jr, Ratanaproeksa AB, Boitano AE, Lucas PC, Castle VP. Constitutively active NFkappa B is required for the survival of S-type neuroblastoma. J Biol Chem 2002;277:42144-50.
- Garkavtsev I, Kozin SV, Chernova O, Xu L, Winkler F, Brown E, et al. The candidate tumour suppressor protein ING4 regulates brain tumour growth and angiogenesis. Nature 2004;428:328-32.
- Brown RE, Tan D, Taylor JS, Miller M, Prichard JW, Kott MM. Morphoproteomic confirmation of constitutively activated mTOR, ERK, and NF-kappaB pathways in high risk neuro-blastoma, with cell cycle and protein analyte correlates. Ann Clin Lab Sci 2007;37:141-7.
- Raychaudhuri B, Han Y, Lu T, Vogelbaum MA. Aberrant constitutive activation of nuclear factor kappaB in glioblastoma multiforme drives invasive phenotype. J Neurooncol 2007. [Epub ahead of print].
- Camandola S, Mattson MP. NF-kappa B as a therapeutic target in neurodegenerative diseases. Expert Opin Ther Targets 2007;11:123-32.
- Zhang CG, Welin D, Novikov L, Kellerth JO, Wiberg M, Hart AM. Motorneuron protection by N-acetyl-cysteine after ventral root avulsion and ventral rhizotomy. Br J Plast Surg 2005;58:765-73.
- Arakawa M, Ishimura A, Arai Y, Kawabe K, Suzuki S, Ishige K, et al. N-Acetylcysteine and ebselen but not nifedipine protected cerebellar granule neurons against 4-hydroxynonenal-induced neuronal death. Neurosci Res 2007;57:220-9.
- Zafarullah M, Li WQ, Sylvester J, Ahmad M. Molecular mechanisms of N-acetylcysteine actions. Cell Mol Life Sci 2003;60:6-20.
- Mihm S, Ennen J, Pessara U, Kurth R, Dröge W. Inhibition of HIV-1 replication and NF-kappa B activity by cysteine and cysteine derivatives. AIDS 1991;5:497-503.
- Schreck R, Rieber P, Baeuerle PA. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J 1991;10:2247-58.
- Shen WH, Zhang CY, Zhang GY. Antioxidants attenuate reperfusion injury after global brain ischemia through inhibiting nuclear factor-kappa B activity in rats. Acta Pharmacol Sin 2003;24:1125-30.
- Muñoz AM, Rey P, Soto-Otero R, Guerra MJ, Labandeira-Garcia JL. Systemic administration of N-acetylcysteine protects dopam inergic neurons against 6-hydroxydopamine-induced degenera-tion. J Neurosci Res 2004;76:551-62.
- Schreck R, Meier B, Männel DN, Dröge W, Baeuerle PA. Dithiocarbamates as potent inhibitors of nuclear factor kappa B activation in intact cells. J Exp Med 1992;175:1181-94.
- Schenk H, Klein M, Erdbrügger W, Dröge W, Schulze-Osthoff K. Distinct effects of thioredoxin and antioxidants on the activation of transcription factors NF-kappa B and AP-1. Proc Natl Acad Sci USA 1994;91:1672-6.
- Ziegler-Heitbrock HW, Sternsdorf T, Liese J, Belohradsky B, Weber C, Wedel A, et al. Pyrrolidine dithiocarbamate inhibits NF-kappa B mobilization and TNF production in human monocytes. J Immunol 1993;151:6986-93.
- Cuzzocrea S, Chatterjee PK, Mazzon E, Dugo L, Serraino I, Britti D, et al. Pyrrolidine dithiocarbamate attenuates the development of acute and chronic inflammation. Br J Pharmacol 2002;135:496-510.
- Nurmi A, Goldsteins G, Närväinen J, Pihlaja R, Ahtoniemi T, Gröhn O, et al. Antioxidant pyrrolidine dithiocarbamate activates Akt-GSK signaling and is neuroprotective in neonatal hypoxia-ischemia. Free Radic Biol Med 2006;40:1776-84.
- Kabay B, Teke Z, Aytekin FO, Yenisey C, Bir F, Sacar M, et al. Pyrrolidine dithiocarbamate reduces lung injury caused by mesenteric ischemia/reperfusion in a rat model. World J Surg 2007;31:1707-15.
- de Jesus Soares T, Costa RS, Balbi AP, Coimbra TM. Inhibition of nuclear factor-kappa B activation reduces glycerol-induced renal injury. J Nephrol 2006;19:439-48.
- Demirbilek S, Akin M, Gürünlüoðlu K, Aydin NE, Emre MH, Tas E, et al. The NF-kappaB inhibitors attenuate hepatic injury in bile duct ligated rats. Pediatr Surg Int 2006;22:655-63.
- Li H, Gu H, Sun B. Protective effects of pyrrolidine dithiocarbamate on myocardium apoptosis induced by adriamycin in rats. Int J Cardiol 2007;114:159-65.
- Crack PJ, Taylor JM, Ali U, Mansell A, Hertzog PJ. Potential contribution of NF-kappaB in neuronal cell death in the glutathione peroxidase-1 knockout mouse in response to ischemia-reperfusion injury. Stroke 2006;37:1533-8.
- Nurmi A, Vartiainen N, Pihlaja R, Goldsteins G, Yrjänheikki J, Koistinaho J. Pyrrolidine dithiocarbamate inhibits translocation of nuclear factor kappa-B in neurons and protects against brain ischaemia with a wide therapeutic time window. J Neurochem 2004;91:755-65. b.
- Chen JW, Chen YH, Lin FY, Chen YL, Lin SJ. Ginkgo biloba extract inhibits tumor necrosis factor-alpha-induced reactive oxygen species generation, transcription factor activation, and cell adhesion molecule expression in human aortic endothelial cells. Arterioscler Thromb Vasc Biol 2003;23:1559-66.
- Levites Y, Youdim MB, Maor G, Mandel S. Attenuation of 6-hydroxydopamine (6-OHDA)-induced nuclear factor-kappaB (NF-kappaB) activation and cell death by tea extracts in neuronal cultures. Biochem Pharmacol 2002;63:21-9.
- Nie G, Cao Y, Zhao B. Protective effects of green tea polyphenols and their major component, (–)-epigallocatechin-3-gallate (EGCG), on 6-hydroxydopamine-induced apoptosis in PC12 cells. Redox Rep 2002;7:171-7.
- Hong JT, Ryu SR, Kim HJ, Lee JK, Lee SH, Yun YP, et al. Protective effect of green tea extract on ischemia/reperfusion-induced brain injury in Mongolian gerbils. Brain Res 2001;888:11-8.
- Lee SY, Lee JW, Lee H, Yoo HS, Yun YP, Oh KW, et al. Inhibitory effect of green tea extract on beta-amyloid-induced PC12 cell death by inhibition of the activation of NF-kappaB and ERK/p38 MAP kinase pathway through antioxidant mechanisms. Brain Res Mol Brain Res 2005;140:45-54.
- Kopp E, Ghosh S. Inhibition of NF-kappa B by sodium salicylate and aspirin. Science 1994;265:956-9.
- Yin MJ, Yamamoto Y, Gaynor RB. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature 1998;396:77-80.
- Grilli M, Pizzi M, Memo M, Spano P. Neuroprotection by aspirin and sodium salicylate through blockade of NF-kappaB activation. Science 1996;274:1383-5.
- Crisanti P, Leon A, Lim DM, Omri B. Aspirin prevention of NMDA-induced neuronal death by direct protein kinase Czeta inhibition. J Neurochem 2005;93:1587-93.
- Maharaj H, Maharaj DS, Daya S. Acetylsalicylic acid and acetaminophen protect against oxidative neurotoxicity. Metab Brain Dis 2006;21:189-99.
- Casper D, Yaparpalvi U, Rempel N, Werner P. Ibuprofen protects dopaminergic neurons against glutamate toxicity in vitro. Neurosci Lett 2000;289:201-4.
- Carrasco E, Werner P. Selective destruction of dopaminergic neurons by low concentrations of 6-OHDA and MPP+: protection by acetylsalicylic acid aspirin. Parkinsonism Relat Disord 2002;8:407-11.
- Esposito E, Di Matteo V, Benigno A, Pierucci M, Crescimanno G, Di Giovanni G. Non-steroidal anti-inflammatory drugs in Parkinson’s disease. Exp Neurol 2007;205:295-312.
- Thomas T, Nadackal TG, Thomas K. Aspirin and non-steroidal anti-inflammatory drugs inhibit amyloid-beta aggregation. Neuroreport 2001;12:3263-7.
- Bisaglia M, Venezia V, Piccioli P, Stanzione S, Porcile C, Russo C, et al. Acetaminophen protects hippocampal neurons and PC12 cultures from amyloid beta-peptides induced oxidative stress and reduces NF-kappaB activation. Neurochem Int 2002;41:43-54.
- Hirohata M, Ono K, Naiki H, Yamada M. Non-steroidal anti-inflammatory drugs have anti-amyloidogenic effects for Alzheimer’s beta-amyloid fibrils in vitro. Neuropharmacology 2005;49:1088-99.
- Tortosa E, Avila J, Pérez M. Acetylsalicylic acid decreases tau phosphorylation at serine 422. Neurosci Lett 2006;396:77-80.
- Natarajan K, Singh S, Burke TR Jr, Grunberger D, Aggarwal BB. Caffeic acid phenethyl ester is a potent and specific inhibitor of activation of nuclear transcription factor NF-kappa B. Proc Natl Acad Sci USA 1996;93:9090-5.
- Amodio R, De Ruvo C, Sacchetti A, Di Santo A, Martelli N, Di Matteo V, et al. Caffeic acid phenethyl ester blocks apoptosis induced by low potassium in cerebellar granule cells. Int J Dev Neurosci 2003;21:379-89.
- Jüttler E, Potrovita I, Tarabin V, Prinz S, Dong-Si T, Fink G, et al. The cannabinoid dexanabinol is an inhibitor of the nuclear factor-kappa B (NF-kappa B). Neuropharmacology 2004;47:580-92.
- Rossi A, Elia G, Santoro MG. Inhibition of nuclear factor kappa B by prostaglandin A1: an effect associated with heat shock transcription factor activation. Proc Natl Acad Sci USA 1997;94:746-50.
- Rossi A, Kapahi P, Natoli G, Takahashi T, Chen Y, Karin M, et al. Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IkappaB kinase. Nature 2000;403:103-8.
- Thomas SC, Ryan MA, Shanley TP, Wong HR. Induction of the stress response with prostaglandin A1 increases I-kappaBalpha gene expression. FASEB J 1998;12:1371-8.
- Castrillo A, Díaz-Guerra MJ, Hortelano S, Martín-Sanz P, Boscá L. Inhibition of IkappaB kinase and IkappaB phosphorylation by 15-deoxy-delta(12,14)-prostaglandin J(2) in activated murine macrophages. Mol Cell Biol 2000;20:1692-8.
- Rovin BH, Lu L, Cosio A. Cyclopentenone prostaglandins inhibit cytokine-induced NF-kappaB activation and chemokine production by human mesangial cells. J Am Soc Nephrol 2001;12:1659-67.
- Siendones E, Fouad D, Díaz-Guerra MJ, de la Mata M, Boscá L, Muntané J. PGE1-induced NO reduces apoptosis by D-galactosamine through attenuation of NF-kappaB and NOS-2 expression in rat hepatocytes. Hepatology 2004;40:1295-303.
- Cernuda-Morollón E, Pineda-Molina E, Cañada FJ, Pérez-Sala D. 15-Deoxy-delta 12,14-prostaglandin J2 inhibition of NF-kappaB-DNA binding through covalent modification of the p50 subunit. J Biol Chem 2001; 276: 35 530–6.
- Straus DS, Pascual G, Li M, Welch JS, Ricote M, Hsiang CH, et al. 15-deoxy-delta 12,14-prostaglandin J2 inhibits multiple steps in the NF-kappa B signaling pathway. Proc Natl Acad Sci USA 2000;97:4844-9.
- Qin ZH, Wang Y, Chen RW, Wang X, Ren M, Chuang DM, et al. Prostaglandin A(1) protects striatal neurons against excitotoxic injury in rat striatum. J Pharmacol Exp Ther 2001;297:78-87.
- Wang X, Qin ZH, Leng Y, Wang Y, Jin X, Chase TN, et al. Prostaglandin A1 inhibits rotenone-induced apoptosis in SH-SY5Y cells. J Neurochem 2002;83:1094-102.
- Zhao X, Zhang Y, Strong R, Grotta JC, Aronowski J. 15d-Prostaglandin J2 activates peroxisome proliferator-activated receptor-gamma, promotes expression of catalase, and reduces inflammation, behavioral dysfunction, and neuronal loss after intracerebral hemorrhage in rats. J Cereb Blood Flow Metab 2006;26:811-20.
- Pereira MP, Hurtado O, Cárdenas A, Boscá L, Castillo J, Dávalos A, et al. Rosiglitazone and 15-deoxy-delta12,14-prostaglandin J2 cause potent neuroprotection after experimental stroke through noncompletely overlapping mechanisms. J Cereb Blood Flow Metab 2006;26:218-29.
- Xu XH, Zhang HL, Han R, Gu ZL, Qin ZH. Enhancement of neuroprotection and heat shock protein induction by combined prostaglandin A1 and lithium in rodent models of focal ischemia. Brain Res 2006;1102:154-62.
- Wayman NS, Hattori Y, McDonald MC, Mota-Filipe H, Cuzzocrea S, Pisano B, et al. Ligands of the peroxisome proliferator-activated receptors (PPAR-gamma and PPAR-alpha) reduce myocardial infarct size. FASEB J 2002;16:1027-40.
- Sun WH, Keller ET, Stebler BS, Ershler WB. Estrogen inhibits phorbol ester-induced IkappaBalpha transcription and protein degradation. Biochem Biophys Res Commun 1998;244:691-5.
- Peet GW, Li J. IkappaB kinases alpha and beta show a random sequential kinetic mechanism and are inhibited by staurosporine and quercetin. J Biol Chem 1999; 274: 32 655–61.
- Kalaitzidis D, Gilmore TD. Transcription factor cross-talk: the estrogen receptor and NF-kappaB. Trends Endocrinol Metab 2005;16:46-52.
- Dikshit P, Goswami A, Mishra A, Chatterjee M, Jana NR. Curcumin induces stress response, neurite outgrowth and prevents NF-kappaB activation by inhibiting the proteasome func-tion. Neurotox Res 2006;9:29-37.
- Lin YZ, Yao SY, Veach RA, Torgerson TR, Hawiger J. Inhibition of nuclear translocation of transcription factor NF-kappa B by a synthetic peptide containing a cell membrane-permeable motif and nuclear localization sequence. J Biol Chem 1995; 270: 14 255–8.
- Liu D, Liu XY, Robinson D, Burnett C, Jackson C, Seele L, Suppression of staphylococcal enterotoxin B-induced toxicity by a nuclear Import inhibitor. J Biol Chem 2004; 279: 19 239–46.
- Chen YL, Law PY, Loh HH. Sustained activation of phosphati-dylinositol 3-kinase/Akt/nuclear factor kappaB signaling mediates G protein-coupled delta-opioid receptor gene expression. J Biol Chem 2006;281:3067-74.
- Qin ZH, Wang Y, Nakai M, Chase TN. Nuclear factor-kappa B contributes to excitotoxin-induced apoptosis in rat striatum. Mol Pharmacol 1998;53:33-42.
- Nakai M, Qin ZH, Chen JF, Wang Y, Chase TN. Kainic acid-induced apoptosis in rat striatum is associated with nuclear factor-kappaB activation. J Neurochem 2000;74:647-58.
- Shou Y, Gunasekar PG, Borowitz JL, Isom GE. Cyanide-induced apoptosis involves oxidative-stress-activated NF-kappaB in cortical neurons. Toxicol Appl Pharmacol 2000;164:196-205.
- Letoha T, Somlai C, Takacs T, Szabolcs A, Jarmay K, Rakonczay Z Jr, et al. A nuclear import inhibitory peptide ameliorates the severity of cholecystokinin-induced acute pancreatitis. World J Gastroenterol 2005;11:990-9.
- Saika S, Miyamoto T, Yamanaka O, Kato T, Ohnishi Y, Flanders KC, et al. Therapeutic effect of topical administration of SN50, an inhibitor of nuclear factor-kappaB, in treatment of corneal alkali burns in mice. Am J Pathol 166:1393-403.
- Ono S, Date I, Onoda K, Shiota T, Ohmoto T, Ninomiya Y, et al. Decoy administration of NF-kappaB into the subarachnoid space for cerebral angiopathy. Hum Gene Ther 1998;9:1003-11.
- De Vry CG, Prasad S, Komuves L, Lorenzana C, Parham C, Le T, et al. Non-viral delivery of nuclear factor-kappaB decoy ameliorates murine inflammatory bowel disease and restores tissue homeostasis. Gut 2007;56:524-33.
- Laguillier C, Hbibi AT, Baran-Marszak F, Metelev V, Cao A, Cymbalista F, et al. Cell death in NF-kappaB-dependent tumour cell lines as a result of NF-kappaB trapping by linker-modified hairpin decoy oligonucleotide. FEBS Lett 2007;581:1143-50.
- Takeuchi K, Itoh H, Yonemitsu Y, Matsumoto T, Kume M, Komori K, et al. In vivo reduction of the nuclear factor-kappaB activity using synthetic cis-element decoy oligonucleotides suppresses intimal hyperplasia in the injured carotid arteries in rabbits. Surg Today 2007;37:575-83.
- Xu MQ, Shuai XR, Yan ML, Zhang MM, Yan LN. Nuclear factor-kappaB decoy oligodeoxynucleotides attenuates ischemia/reperfusion injury in rat liver graft. World J Gastroenterol 2005;11:6960-7.
- Matsuda N, Hattori Y, Takahashi Y, Nishihira J, Jesmin S, Kobayashi M, et al. Therapeutic effect of in vivo transfection of transcription factor decoy to NF-kappaB on septic lung in mice. Am J Physiol Lung Cell Mol Physiol 2004;287:L1248-55.
- Kawamura I, Morishita R, Tsujimoto S, Manda T, Tomoi M, Tomita N, et al. Intravenous injection of oligodeoxynucleotides to the NF-kappaB binding site inhibits hepatic metastasis of M5076 reticulosarcoma in mice. Gene Ther 2001;8:905-12.
- Jun-Ichi S, Hiroshi I, Ryo G, Ryuichi M, Kensuke E, Mitsuaki I. Initial clinical cases of the use of a NF-kappaB decoy at the site of coronary stenting for the prevention of restenosis. Circ J 2004;68:270-1.
- Isomura I, Morita A. Regulation of NF-kappaB signaling by decoy oligodeoxynucleotides. Microbiol Immunol 2006;50:559-63.
- Nakagami H, Tomita N, Kaneda Y, Ogihara T, Morishita R. Anti-oxidant gene therapy by NF kappa B decoy oligodeoxy-nucleotide. Curr Pharm Biotechnol 2006;7:95-100.
- Sharma RK, Garg BS, Kurosaki H, Goto M, Otsuka M, Yamamoto T, et al. Aurine tricarboxylic acid, a potent metal-chelating inhibitor of NFkappaB-DNA binding. Bioorg Med Chem 2000;8:1819-23.
- Terai K, Matsuo A, McGeer EG, McGeer PL. Enhancement of immunoreactivity for NF-kappa B in human cerebral infarctions. Brain Res 1996;739:343-9.
- Clemens JA, Stephenson DT, Dixon EP, Smalstig EB, Mincy RE, Rash KS, et al. Global cerebral ischemia activates nuclear factor-kappa B prior to evidence of DNA fragmentation. Brain Res Mol Brain Res 1997;48:187-96.
- Stephenson D, Yin T, Smalstig EB, Hsu MA, Panetta J, Little S, et al. Transcription factor nuclear factor-kappa B is activated in neurons after focal cerebral ischemia. J Cereb Blood Flow Metab 2000;20:592-603.
- Schwaninger M, Inta I, Herrmann O. NF-kappaB signalling in cerebral ischaemia. Biochem Soc Trans 2006;34:1291-4.
- Clemens JA, Stephenson DT, Yin T, Smalstig EB, Panetta JA, Little SP. Drug-induced neuroprotection from global ischemia is associated with prevention of persistent but not transient activation of nuclear factor-kappaB in rats. Stroke 1998;29:677-82.
- Nurmi A, Lindsberg PJ, Koistinaho M, Zhang W, Juettler E, Karjalainen-Lindsberg ML, et al. Nuclear factor-kappaB contributes to infarction after permanent focal ischemia. Stroke 2004;35:987-91. a.
- Zhang HL, Huang ZH, Zhu Y, Liang ZQ, Han R, Wang XX, et al. Neuroprotective effects of prostaglandin A1 in animal models of focal ischemia. Brain Res 2005;1039:203-6.
- Schneider A, Martin-Villalba A, Weih F, Vogel J, Wirth T, Schwaninger M. NF-kappaB is activated and promotes cell death in focal cerebral ischemia. Nat Med 1999;5:554-9.
- Hunot S, Brugg B, Ricard D, Michel PP, Muriel MP, Ruberg M, et al. Nuclear translocation of NF-kappaB is increased in dopaminergic neurons of patients with parkinson disease. Proc Natl Acad Sci USA 1997;94:7531-6.
- Dehmer T, Heneka MT, Sastre M, Dichgans J. Protection by pioglitazone in the MPTP model of Parkinson’s disease correlates with IκBα induction and block of NF kappa B and iNOS activation. J Neurochem 2004;88:494-501.
- Terai K, Matsuo A, McGeer PL. Enhancement of immunoreactivity for NF-kappa B in the hippocampal formation and cerebral cortex of Alzheimer’s disease. Brain Res 1996;735:159-68.
- Boissière F, Hunot S, Faucheux B, Duyckaerts C, Hauw JJ, Agid Y, et al. Nuclear translocation of NF-kappaB in cholinergic neurons of patients with Alzheimer’s disease. Neuroreport 1997;8:2849-52.
- Ferrer I, Martí E, López E, Tortosa A. NF-κB immunoreactivity is observed in association with beta A4 diffuse plaques in patients with Alzheimer’s disease. Neuropathol Appl Neurobiol 1998;24:271-7.
- Kitamura Y, Shimohama S, Ota T, Matsuoka Y, Nomura Y, Taniguchi T. Alteration of transcription factors NF-kappaB and STAT1 in Alzheimer’s disease brains. Neurosci Lett 1997;237:17-20.
- Lukiw WJ, Bazan NG. Strong nuclear factor-kappaB-DNA binding parallels cyclooxygenase-2 gene transcription in aging and in sporadic Alzheimer’s disease superior temporal lobe neocortex. J Neurosci Res 1998;53:583-92.
- Huang Y, Liu F, Grundke-Iqbal I, Iqbal K, Gong CX. NF-kappaB precursor, p105, and NF-kappaB inhibitor, IkappaBgamma, are both elevated in Alzheimer disease brain. Neurosci Lett 2005;373:115-8.
- Kaltschmidt B, Uherek M, Volk B, Baeuerle PA, Kaltschmidt C. Transcription factor NF-kappaB is activated in primary neurons by amyloid beta peptides and in neurons surrounding early plaques from patients with Alzheimer disease. Proc Natl Acad Sci USA 1997;94:2642-7.
- Bales KR, Du Y, Dodel RC, Yan GM, Hamilton-Byrd E, Paul SM. The NF-kappaB/Rel family of proteins mediates Abeta-induced neurotoxicity and glial activation. Brain Res Mol Brain Res 1998;57:63-72.
- Samuelsson M, Fisher L, Iverfeldt K. beta-Amyloid and interleukin-1beta induce persistent NF-kappaB activation in rat primary glial cells. Int J Mol Med 2005;16:449-53.
- Kaltschmidt B, Uherek M, Wellmann H, Volk B, Kaltschmidt C. Inhibition of NF-kappaB potentiates amyloid beta-mediated neuronal apoptosis. Proc Natl Acad Sci USA 1999;96:9409-14.
- Paris D, Patel N, Quadros A, Linan M, Bakshi P, Ait-Ghezala G, et al. Inhibition of Abeta production by NF-kappaB inhibitors. Neurosci Lett 2007;415:11-6.
- Valerio A, Boroni F, Benarese M, Sarnico I, Ghisi V, Bresciani LG, et al. NF-kappaB pathway: a target for preventing beta-amyloid (Abeta)-induced neuronal damage and Abeta42 production. Eur J Neurosci 2006;23:1711-20.
- Imbimbo BP. The potential role of non-steroidal anti-inflammatory drugs in treating Alzheimer’s disease. Expert Opin Investig Drugs 2004;13:1469-81.
- Kaltschmidt C, Kaltschmidt B, Baeuerle PA. Stimulation of ionotropic glutamate receptors activates transcription factor NF-kappa B in primary neurons. Proc Natl Acad Sci USA 1995;92:9618-22.
- de Erausquin GA, Hyrc K, Dorsey DA, Mamah D, Dokucu M, Mascó DH, et al. Nuclear translocation of nuclear transcription factor-kappa B by alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptors leads to transcription of p53 and cell death in dopaminergic neurons. Mol Pharmacol 2003;63:784-90.
- Zou J, Crews F. CREB and NF-kappaB transcription factors regulate sensitivity to excitotoxic and oxidative stress induced neuronal cell death. Cell Mol Neurobiol 2006;26:385-405.
- Cherng JM, Lin HJ, Hung MS, Lin YR, Chan MH, Lin JC. Inhibition of nuclear factor kappaB is associated with neuroprotective effects of glycyrrhizic acid on glutamate-induced excitotoxicity in primary neurons. Eur J Pharmacol 2006;547:10-21.
- Nakai M, Qin ZH, Wang Y, Chase TN. Free radical scavenger OPC-14117 attenuates quinolinic acid-induced NF-kappaB activation and apoptosis in rat striatum. Brain Res Mol Brain Res 1999;64:59-68.
- Wang Y, Qin ZH, Nakai M, Chen RW, Chuang DM, Chase TN. Co-stimulation of cyclic-AMP-linked metabotropic glutamate receptors in rat striatum attenuates excitotoxin-induced nuclear factor-kappaB activation and apoptosis. Neuroscience 1999;94:1153-62.
- Lipton SA. Janus faces of NF-kappa B: neurodestruction versus neuroprotection. Nat Med 1997;3:20-2.
- Barkett M, Gilmore TD. Control of apoptosis by Rel/NF-kappaB transcription factors. Oncogene 1999;18:6910-24.
- Kaltschmidt B, Baeuerle PA, Kaltschmidt C. Potential involvement of the transcription factor NF-kappa B in neurological disorders. Mol Aspects Med 1993;14:171-90.
- Pizzi M, Spano P. Distinct roles of diverse nuclear factor-kappaB complexes in neuropathological mechanisms. Eur J Pharmacol 2006;545:22-8.