
Hydrogen sulfide: third gaseous transmitter, but with great pharmacological potential
Introduction
For hundreds of years, hydrogen sulfide (H2S) has been known solely as a toxic gas with the smell of rotten eggs. Indeed, H2S is gradually considered to be a broad spectrum toxicant; its major toxic effects are the toxicity of the central nervous system (CNS) and the inhibition of the respiratory system[1,2]. Recent studies have shown that H2S is not only a chemical hazard in certain industrial manufacturing, but it can also be produced endogenously in mammalian tissues from L-cysteine mainly by 3 enzymes: cystathionine β-synthetase (CBS), cystathionine γ-lyase (CSE), and 3-mercapto-sulfurtransferase[3–6]. The expressions of CBS and CSE have been detected in various tissues. H2S research has been recently focused on its physiological effects and significance under disease conditions. Like nitric oxide (NO) and carbon monoxide (CO), which are considered 2 gaseous transmitters, H2S has been shown to be the third gaseous transmitter and plays important roles, both in normal physiology conditions and in the process/progress of several diseases.
H2S is a small molecule and can permeate membranes freely. The endogenous H2S level is 0–46 µmol/L[7] in rat serum and 50–160 µmol/L in the brain[4]. One-third of H2S remains undissociated in an aqueous solution and its solubility in lipophilic solvents is 5-fold greater than in water[8]. H2S is mostly metabolized to sulfate and thiosulfate via the oxidation metabolism in mitochondria, and glutathione triggers the reaction. Very little of H2S can be converted into lower toxic compounds of methylmercaptan and dimethyl sulfate via the methylation metabolism in cytosol. The metabolic product can exhaust from the kidney and intestinal tract and lungs within 24 h, so the endogenously-generated H2S under physiological condition is hardly accumulated or toxic to cells due to the balanced cellular metabolism of the gas[9].
Although there are several excellent reviews on the pathophysiological effects of H2S, such as those by Wang[8] in 2002, Tang et al[6] and Du et al[10] in 2006, the present study will summarize the latest progress on H2S studies concerning the CNS, cardiovascular system, a possible novel mechanism for its motor effect, as well as the role of H2S in ischemia.
Role of H2S in the CNS
Endogenous H2S is generated mainly by CBS in the brain[11]. The transcriptional expression of CBS, but not CSE, in the rat brain (hippocampus, cerebellum, cerebral, and brainstem) was confirmed using the Northern blot assay; a similar conclusion was confirmed in CBS knockout mice. The production of H2S by CBS in the brain is regulated by Ca2+ and calmodulin[12]. It is greatly enhanced by the activation of glutamate receptors and electrical stimulation which could cause influx of Ca2+. There are 2 mechanisms through which CBS could produce H2S. First, CBS could catalyze the production of H2S from cysteine by a β-elimination or a α,β-elimination reaction; second, CBS can efficiently catalyze the formation of H2S via the condensation of homocysteine with cysteine, and the latter is not affected by Ca2+ and calmodulin[13]. The regulation of CBS activity can affect brain H2S formation. Eto et al found that sodium nitroprusside (SNP), well known as a NO donor, could enhance CBS activity by modifying 4 of 13 cysteine residues of CBS, but this effect is independent of NO production[14]. A human CBS complete genomic sequence has been determined[15] and the transcriptional start sites of 5 CBS mRNA isoforms, designated CBS-1a–1e, have been mapped[16]. The 5 CBS transcripts begin with a different exon. CBS gene transcription might be regulated by more than 1 promoter. Isoforms -1a and -1b form the vast majority of transcripts. Regulation on the transcriptional level is likely to be the mechanism of the tissue-specific manner of CBS expression, and in some senses, the expression of CBS in the brain could be modulated at the gene level under physiological and pathophysiological conditions.
The endogenous H2S by CBS in the brain indicates it has physiological functions in the CNS. N-methyl-D-aspartate (NMDA) receptors may be one of its targets. The activation of NMDA receptors is required for the induction of hippocampal long-term potentiation (LTP)[17], a synaptic model of learning and memory. Because of the relatively high concentration of endogenous H2S in the brain (50–160 µmol/L), the physiological concentration of H2S facilitates the induction of LTP by enhancing NMDA receptor-induced currents[11]. This activation could be blocked by an adenylyl cyclase-specific inhibitor, indicating that the modulation of NMDA receptors by H2S is induced by the enhancement of cAMP production[18]. This function ranked H2S as a neuromodulator in the brain. Another study showed that H2S could increase intracellular Ca2+ and induce Ca2+ waves in neighboring astrocytes[19]. Therefore, H2S may mediate signals between neurons and glia and regulate synaptic activity by modulating the activity of both neurons and glia.
In addition to its role in signal transduction, H2S can protect neuron cells from oxidative stress, not only by increasing the levels of antioxidant glutathione[20], but also by activating the K+ATP and Cl– channels[21]. In human cultured neuron cells, H2S could inhibit peroxynitrite (ONOO–), which is an important mediator of human neurodegenerative disease, inducing tyrosine nitration, a1-antiproteinase inactivation, cell toxicity, intracellular protein oxidation, and protein nitration. This antioxidant action of H2S suggests it functions as an endogenous ONOO– scavenger[22]. Oxidative stress is responsible for neuronal damage and degenerates in brain disorders. These observations suggest that H2S may act as a neuroprotectant against oxidative stress.
The concentration of H2S in the brain changes with CNS diseases. The levels of H2S decreased by 55% in the brains of Alzheimer’s disease (AD) patients and CBS activity was also dramatically decreased[23], but the level of AdoMet, a CBS activator, is low in AD brains. The CBS activity was reduced in another disease, homocystinuria[24]. Febrile seizure (FS) frequently occurs in children. Both gamma-aminobutyric acid (GABA) B receptor (GABABR) subunits and the H2S/CBS system were involved in FS. H2S functioned as a protective factor in the development of FS through regulating GABABR[25]. Intriguingly, the levels of CBS in Down’s syndrome brains were approximately 3 times greater than those in normal individuals[26]. The role of H2S in CNS diseases is not clear and needs to be explored in future. The progress of this field can provide novel therapy in clinical trials.
Role of H2S in the cardiovascular system
Hosoki et al found that H2S could be generated in the homogenates of the portal vein and thoracic aorta[4]. They also identified that CSE was the major enzyme to generate H2S in these tissues by detecting the transcription of the mRNA of CSE with the Northern blot assay. The expression levels of CSE mRNA varied in different types of vascular tissues and was ranked as artery>aorta>tail artery>mesenteric artery[27]. A recent study has shown that in the heart, there is very few CBS, but plentiful CSE[28]. It seems that CBS does not play a major role in the cardiovascular system under physiological conditions. These observations suggested the potential physiological functions of H2S/the CSE system in the cardiovascular system. The biosynthetic underlying mechanism of action of H2S is summarized in Figure 1[3].
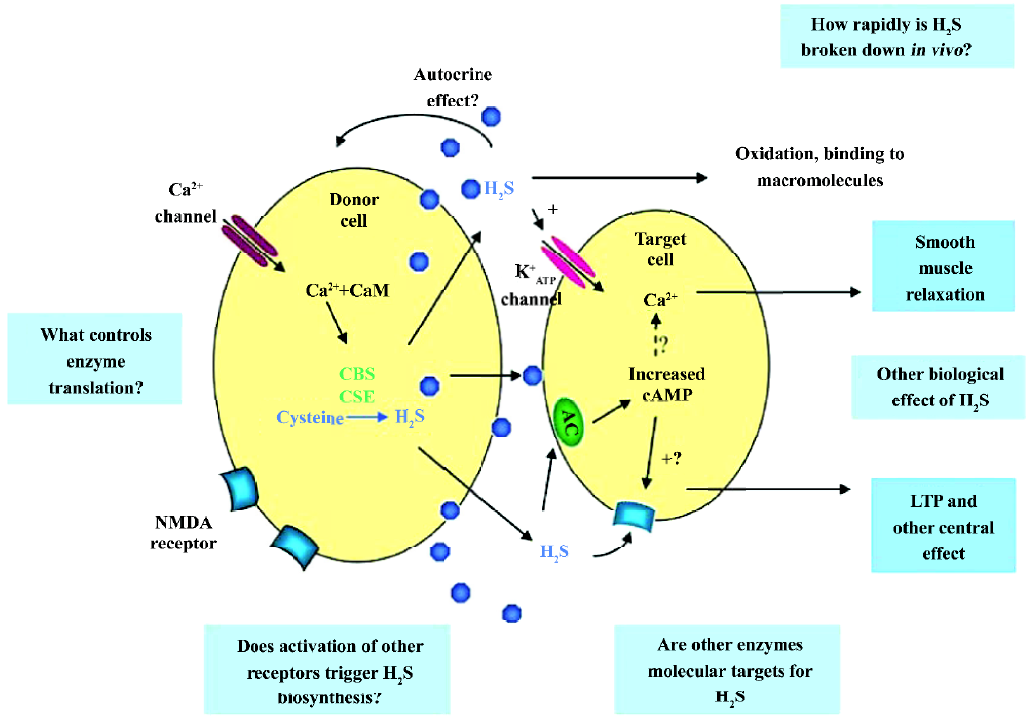
H2S can be produced enzymatically in vascular tissues and relaxes vascular smooth muscles both in vivo and in vitro[4,29]. This vasorelaxant effect is most probably caused by opening vascular smooth muscle cells (VSMC) KATP channels which leads to membrane hyperpolarization[27]. Therefore, H2S may reduce extracellular Ca2+ entry and relax vascular tissues. The vasorelaxation induced by H2S can be attenuated by the removal of the endothelium, since H2S may facilitate the release of vasorelaxant factors from the endothelium, including NO and the endothelium-derived relaxing factor. As opposed to NO and CO, H2S-induced vasorelaxation is not mediated by the cGMP signaling pathway. This indicates that H2S is a novel endogenous gaseous modulator of vascular contractility. At the same time, similar to NO and CO, H2S could inhibit VSMC proliferation and induce apoptosis in vitro[30,31]. Using cultured VSMC, exogenous H2S could dose-dependently suppress the proliferation of VSMC through the mitogen-activated protein kinase (MAPK) signaling pathway. Studies using molecular means to overexpress CSE in cultured VSMC found that endogenous H2S could also attenuate the rate of cell proliferation and increase the rate of cell apoptosis. The effect is via the activation of MAPK and caspase-3. The possible signaling pathway is shown in Figure 2. So H2S is not only a vasorelaxant, but also an important regulator of cell growth and may thereby attenuate the structural remodeling of vessel tissues. This can help us understand the mechanism of some vascular diseases and provide links to new therapeutic methods.
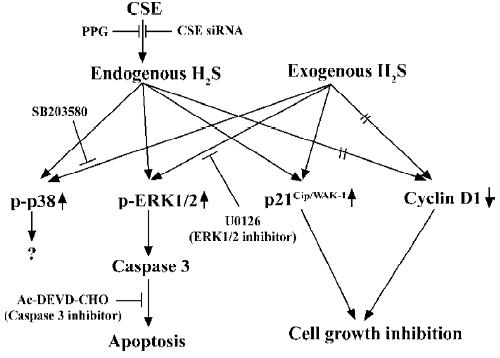
The pathophysiological role of H2S in some cardiovascular diseases has been explored. The endogenous H2S/CSE pathway participated in the pathophysiological process in vascular diseases, such as spontaneous hypertension[32], hypoxia-induced pulmonary hypertension (HPH)[33–35], and high pulmonary blood flow-induced pulmonary hypertension[36,37]. Hypertension is one of the most common cardiovascular diseases and its mechanism is not fully understood yet. Vasoconstriction and structural remodeling by VSMC proliferation are essential processes in hypertension develop-ment. In the spontaneous hypertension rat model, the plasma level of H2S is low and the activity of CSE is suppressed. The exogenous administration of H2S not only attenuated the elevation of blood pressure and vessel remodeling, but also recovered the H2S level and CSE activity[32]. Guang et al’s study found that there was dysfunction of the H2S/CSE system in L-NAME-induced hypertension rats and that exogenous H2S could effectively prevent the development of hypertension[38]. The exogenous H2S-induced positive feedback on CSE activity is different from its negative feedback in physiological conditions[39]. A recent study reported that the exogenous administration of H2S downregulated osteopontin gene expression and ameliorated vascular calcification (which is a common finding in many diseases, such as hypertension, atherosclerosis, diabetes, chronic renal failure, aging, and arterial stenosis)[40]. As it is known, the baroreflex is the major method of blood pressure modulation. Exogenous H2S could facilitate carotid sinus baroreflex (CSB) by opening KATP channels and further closing the calcium channels in vascular smooth muscle which suggests that endogenous H2S might activate the activity of the CSB in vivo[41]. In HPH pathophysiological processes, the similar dysfunction of H2S/CSE was found, and exogenous H2S could inhibit the proliferative cell nuclear antigen (PCNA) and U-II expressions in the pulmonary wall to depress the proliferation of pulmonary artery smooth muscle cells and reduce the expression of collagen I and III, elastin, and TGF-β3 to decrease the hypoxic pulmonary vascular structural remodeling[34,35]. Olson et al’s recent study on the mechanical and electrical responses of select blood vessels to hypoxia and H2S suggested that H2S served as an O2 sensor/transducer in the vascular responses to hypoxia. The inhibition of H2S synthesis inhibited the hypoxic response of vertebrate blood vessels and the concentration of H2S in the vessel was regulated by the balance between endogenous H2S production and its oxidation by available O2[42]. The exogenous supply of H2S could alleviate the elevation of pulmonary arterial pressure. At the same time, exogenous supply of propargylglycine (PPG, inhibitor of CSE), plasma CO level, and the expressions of the HO-1 protein and mRNA in pulmonary arteries decreased. The results showed that H2S could play a regulatory role in the pathogenesis of HPH through the upregulation of the CO/HO pathway[43]. How-ever, in aortic smooth muscle cells, Jin et al’s study proved that endogenous CO/HO and the H2S/CSE pathways downregulated each other under physiological conditions[44]. Exogenous H2S also ameliorated pulmonary vascular structural remodeling induced by high pulmonary blood flow, downregulated PCNA expression and the ERK/MAPK signal pathway, inhibited the NO/NO synthase pathway, and enhanced the CO/HO pathway in rats with high pulmonary blood flow[36,37]. These studies suggested that endogenous H2S was one of the key factors in hypertension development and that the deficit of the H2S/CSE system was one of the major causes of hypertension.
Some studies have proven that the concentration of arterial endogenous H2S was significantly increased in both septic and endotoxic shock rats, which suggested that endogenous H2S was still involved in physiological and pathophysiological processes during shock[45].
Human cystathioninuria, which is characterized by high plasma homocysteine and cystathionine, is concerned with a wide range of disease associations, such as cardiovascular injury. Its genomic basis has been shown to be 2 nonsense mutations and 2 sense mutations in CSE[46].
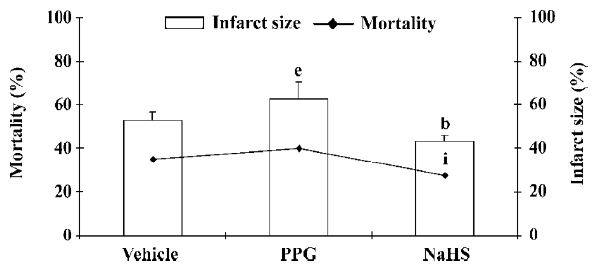
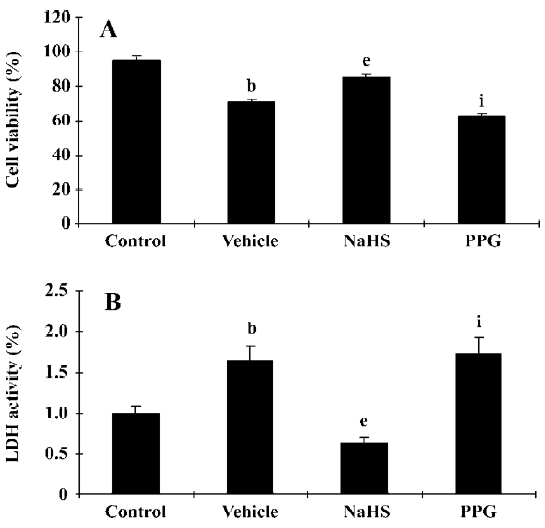
It has been reported that H2S also activates the KATP channel in mitochondria and sarcolemmal KATP channels in cardiac myocytes and has potent cardioprotective effects[47–49]. The cardioprotection of H2S was also demonstrated in rat isolated ventricular myocytes[50]. NaHS could concentration-dependently increase the cell viability and the percentage of rod-shaped cells, which were exposed to severe metabolic inhibition solution. NaHS-induced cardioprotection following metabolic inhibition preconditioning could be blocked by PPG and HMR-1098 (a sarcolemmal KATP blocker). Pretreatment with L-NAME to block endogenous NO production could also attenuate the cardioprotective effect of NaHS. These results indicate that H2S may protect the heart most probably by activating sarcolemmal KATP, which is different from its vasorelaxant effect. NO also plays an important role in cardioprotection. Further, the cardioprotective effect has similar characteristics to the time-course of ischemic preconditioning; it suggests the possible protective role of H2S in ischemic myocardium. This has been proven by a recent study[51]. In a rat model of myocardial infarction (MI), NaHS treatment could decrease the mortality rate of MI rats and diminish infarct size as shown in Figure 3. The vessel dilating/relaxing effects of NaHS may dilate coronary arteries and increase coronary blood flow in ischemic diseases, thus reducing cellular damage from ischemia. This heart protective effect could be abolished by the administration of PPG. The results were further confirmed by our in vitro hypoxic model (Figure 4). It has been suggested that endogenous H2S might provide a novel approach to the treatment of MI. Further work needs to be performed to explore whether or not the mechanism of this effect concerns the KATP channel. H2S also showed a cardio-protective effect in another isoproterenol injection-induced myocardial ischemic injury model in which the plasma H2S concentration and CSE activity decreased. The administration of NaHS could effectively protect myocytes and contractile activity[52].
Excitatory motor effect of H2S
In contrast to the vasorelaxant effect, NaHS produced concentration-dependent contractile responses in the detrusor muscle of the rat urinary bladder[53]. This response exhibited rapid and persistent tachyphylaxis similar to the responses of capsaicin[54,55]. The response was abolished by high-capsaicin pretreatment which could desensitize capsaicin-sensitive primary afferent neurons or the pretreatment of tissues with a combination of tachykinin natural killer (NK)1 and NK2 receptor-selective antagonists. At the same time, the response to NaHS is mostly resistant to tetrodotoxin, as is the effect of capsaicin in this organ[56]. These results show pharmacological evidence that H2S stimulates capsaicin-sensitive primary afferent nerve terminals with the consequent release of tachykinins, which in turn produces contractile responses of the detrusor muscle. In further studies, the same researchers demonstrated that the transient receptor potential vanilloid receptor 1 (TRPV1, also called the capsaicin receptor) selective antagonist capsazepine and SB366791 could not affect the H2S contractile activity[53]. However, the unselective cation channel blocker, ruthenium red, almost abolished the contraction similar to its effect on capsaicin, which provided 2 hypotheses: first, H2S stimulates the TRPV1 receptor by a different way from those known activators; second, H2S might stimulate other receptors present on the terminals of capsaicin-sensitive sensory neurons.
TRPV1 was cloned from rat sensory neurons[57] in 1997. TRPV1 is non-selective cation channel with high permeability of Ca2+ and could be activated by chemical and physical stimuli, such as capsaicin, low pH[58], noxious heat, ananda-mide[59], 12-hydroperoxyeicosatetraenoic acid[60], and N-arachidonoyl-dopamine[61]. The influx of Na+ causes primary sensory neuron depolarization and the initiation of action potentials. In particular, the influx of Ca2+ resulted in the local release of neuropeptides, including the calcitonin gene-related peptide and the tachykinins, substance P, and neurokinin A. TRPV1-positive neurons are not only afferent neurons which are involved in the perception of somatic and visceral pain, but also have a sensory effector function. These neuropeptides act on different effector cells and cause different responses, including neurogenic inflammation, thermal hyperalgesia, airway constriction, and vasodilatation[62]. TRPV1 is highly expressed in primary sensory neurons of the trigeminal, vagal, and dorsal root ganglion with C- and A-δ fibers, which are called nociceptive neuron. Studies also show that TRPV1 is expressed in non-neuronal cells, including epithelial cells of the urothelium[63], keratinocytes[64], and skeletal muscles[65]. These features of TRPV1 indicate its broad physiological and pathophysiological functions.
In addition to the detrusor muscle, NaHS increases sensory neuropeptide release in the guinea pig airways and causes in vivo bronchoconstriction and microvascular leakage in a capsazepine-sensitive manner[66]. This novel mechanism may contribute to the irritant action of H2S in the respiratory system, possibly through TRPV1 activation. Further research is still required in order to prove whether or not H2S acts as a endogenous ligand of TRPV1. It will be interesting to detect the odorous activator of TRPV1 and its undergoing mechanism. Therefore, previous studies on the physiological and pathophysiological roles of H2S need re-evaluation based on this mechanism.
Concluding remarks
H2S has been shown to be an important biological molecule in the last 2 decades. In addition to its neuromodulator and cardiovascular protection effects, studies also show that H2S has various effects in mammalian tissues, such as the relaxation effect of the ileum, which indicates that endogenous H2S could regulate alimentary contractile functions[67]. H2S has been described as an endogenous mediator with diverse biological effects in a study, including playing an important role in endotoxin-induced inflammation[68]. The studies of the physiological functions of H2S and its underlying mechanism, the regulation of H2S concentration and activity, and/or the expression of CBS and/or CSE and its interaction with some diseases may have a significant impact in our understanding of the pathogenesis of these diseases, as well as having far-reaching clinical and therapeutic implications. Further research needs to be undertaken to find new therapeutic methods and ruling out possible side-effects.
References
- Beauchamp RJ, Bus JS, Popp JA, Boreiko CJ, Andjelkovich DA. A critical review of the literature on hydrogen sulfide toxicity. Crit Rev Toxicol 1984;13:25-97.
- Guidotti TL. Hydrogen sulphide. Occup Med (Lond) 1996;46:367-71.
- Moore PK, Bhatia M, Moochhala S. Hydrogen sulfide: from the smell of the past to the mediator of the future? Trends Pharmacol Sci 2003;24:609-11.
- Hosoki R, Matsuki N, Kimura H. The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem Biophys Res Commun 1997;237:527-31.
- Kimura H, Nagai Y, Umemura K, Kimura Y. Physiological roles of hydrogen sulfide: synaptic modulation, neuroprotection, and smooth muscle relaxation. Antioxid Redox Signal 2005;7:795-803.
- Tang CS, Li XH, Du JB. Hydrogen sulfide as a new endogenous gaseous transmitter in the cardiovascular system. Curr Vasc Pharmacol 2006;4:17-22.
- Zhao W, Ndisang JF, Wang R. Modulation of endogenous production of H2S in rat tissues. Can J Physiol Pharmacol 2003;81:848-53.
- Wang R. Two’s company, three’s a crowd: can H2S be the third endogenous gaseous transmitter? FASEB J 2002;16:1792-8.
- Furne J, Springfield J, Koenig T, DeMaster E, Levitt MD. Oxidation of hydrogen sulfide and methanethiol to thiosulfate by rat tissues: a specialized function of the colonic mucosa. Biochem Pharmacol 2001;62:255-9.
- Du JB, Zhang CY, Yan H, Tang CS. A newly found gasotransmitter, hydrogen sulfide, in the pathogenesis of hypertension and other cardiovascular diseases. Curr Hypertens Rev 2006;2:123-6.
- Abe K, Kimura H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J Neurosci 1996;16:1066-71.
- Eto K, Kimura H. The production of hydrogen sulfide is regulated by testosterone and S-adenosyl-L-methionine in mouse brain. J Neurochem 2002;83:80-6.
- Chen X, Jhee KH, Kruger WD. Production of the neuromodulator H2S by cystathionine beta-synthase via the condensation of cysteine and homocysteine. J Biol Chem 2004;279:52082-6.
- Eto K, Kimura H. A novel enhancing mechanism for hydrogen sulfide-producing activity of cystathionine beta-synthase. J Biol Chem 2002;277:42680-5.
- Avila MA, Berasain C, Torres L, Martin-Duce A, Corrales FJ, Yang H, et al. Reduced mRNA abundance of the main enzymes involved in methionine metabolism in human liver cirrhosis and hepatocellular carcinoma. J Hepatol 2000;33:907-14.
- Bao L, Vlcek C, Paces V, Kraus JP. Identification and tissue distribution of human cystathionine beta-synthase mRNA isoforms. Arch Biochem Biophys 1998;350:95-103.
- Harris EW, Ganong AH, Cotman CW. Long-term potentiation in the hippocampus involves activation of N-methyl-D-aspartate receptors. Brain Res 1984;323:132-7.
- Kimura H. Hydrogen sulfide induces cyclic AMP and modulates the NMDA receptor. Biochem Biophys Res Commun 2000;267:129-33.
- Nagai Y, Tsugane M, Oka J, Kimura H. Hydrogen sulfide induces calcium waves in astrocytes. FASEB J 2004;18:557-9.
- Kimura Y, Kimura H. Hydrogen sulfide protects neurons from oxidative stress. FASEB J 2004;18:1165-7.
- Kimura Y, Dargusch R, Schubert D, Kimura H. Hydrogen sulfide protects HT22 neuronal cells from oxidative stress. Antioxid Redox Signal 2006;8:661-70.
- Whiteman M, Cheung NS, Zhu YZ., Chu SH, Siau JL, Wong BS, et al. Hydrogen sulphide: a novel inhibitor of hypochlorous acid-mediated oxidative damage in the brain? Biochem Biophys Res Commun 2005;326:794-8.
- Eto K, Asada T, Arima K, Makifuchi T, Kimura H. Brain hydrogen sulfide is severely decreased in Alzheimer’s disease. Biochem Biophys Res Commun 2002;293:148-58.
- Hu FL, Gu Z, Kozich V, Kraus JP, Ramesh V, Shih VE. Molecular basis of cystathionine beta-synthase deficiency in pyridoxine responsive and nonresponsive homocystinuria. Hum Mol Genet 1993;2:1857-60.
- Han Y, Qin J, Bu DF, Chang XZ, Yang ZX, Du JB. Gamma-aminobutyric acid B receptor regulates the expression of hydrogen sulfide/cystathionine-beta-synthase system in recurrent febrile seizures. Zhongguo Dang Dai Er Ke Za Zhi 2006;8:141-3. Chinese..
- Ichinohe A, Kanaumi T, Takashima S, Enokido Y, Nagai Y, Kimura H. Cystathionine beta-synthase is enriched in the brains of Down’s patients. Biochem Biophys Res Commun 2005;338:1547-50.
- Zhao W, Zhang J, Lu Y, Wang R. The vasorelaxant effect of H2S as a novel endogenous gaseous K(ATP) channel opener. EMBO J 2001;20:6008-16.
- Geng B, Yan H, Zhong GZ, Zhang CY, Chen XB, Jiang HF, et al. Hydrogen sulfide: a novel cardiovascular functional regulatory gas factor. Beijing Da Xue Xue Bao 2004;36:106. Chinese..
- Zhao W, Wang R. H. (2)S-induced vasorelaxation and underlying cellular and molecular mechanisms. Am J Physiol Heart Circ Physiol 2002;283:474-80.
- Du JB, Yan H, Cheung YF, Geng B, Jiang HF, Chen XB, et al. The possible role of hydrogen sulfide as a smooth musle cell proliferation inhibitor in rat cultured cells. Heart Vessels 2004;19:75-80.
- Yang G, Sun X, Wang R. Hydrogen sulfide-induced apoptosis of human aorta smooth muscle cells via the activation of mitogen-activated protein kinases and caspase-3. FASEB J 2004;18:1782-4.
- Yan H, Du JB, Tang CS. The possible role of hydrogen sulfide on the pathogenesis of spontaneous hypertension in rats. Biochem Biophys Res Commun 2004;313:22-7.
- Zhang C, Du J, Bu DB, Yan H, Tang X, Si Q, et al. The regulatory effect of endogenous hydrogen sulfide on hypoxic pulmonary hypertension. Beijing Da Xue Xue Bao 2003;35:488-93. Chinese..
- Li XH, Du JB, Shi L, Li J, Tang XY, Qi JG, et al. Down-regulation of endogenous hydrogen sulfide pathway in pulmonary hypertension and pulmonary vascular structural remodeling induced by high pulmonary blood flow in rats. Circ J 2005;69:1418-24.
- Jin HF, Cong BL, Zhao B, Zhang CY, Liu XM, Zhou WJ, et al. Effects of hydrogen sulfide on hypoxic pulmonary vascular structural remodeling. Life Sci 2006;78:1299-309.
- Wang YF, Shi L, Du JB, Tang CS. Impact of L-arginine on hydrogen sulfide/cystathionine-γ-lyase pathway in rats with high blood flow-induced pulmonary hypertension. Biochem Biophys Res Commun 2006;345:851-7.
- Li XH, Du JB, Bu DF, Tang XY, Tang CS. Sodium hydrosulfide alleviated pulmonary vascular structural remodeling induced by high pulmonary blood flow in rats. Acta Pharmacol Sin 2006;27:977-86.
- Zhong GZ, Chen FR, Cheng YQ, Tang CS, Du JB. The role of hydrogen sulfide in the pathogenesis of hypertension in rats induced by inhibition of nitric oxide synthase. J Hypertens 2003;21:1879-85.
- Kredich NM, Foote LJ, Keenan BS. The stoichiometry and kinetics of the inducible cysteine desulfhydrase from Salmonella typhimurium. J Biol Chem 1973;248:6187-96.
- Wu SY, Pan CS, Geng B, Zhao J, Yu F, Pang YZ, et al. Hydrogen sulfide ameliorates vascular calcification induced by vitamin D3 plus nicotine in rats. Acta Pharmacol Sin 2006;27:299-306.
- Xiao L, Wu YM, Zhang H, Liu YX, He RR. Hydrogen sulfide facilitates carotid sinus baroreflex in anesthetized rats. Acta Pharmacol Sin 2006;27:294-8.
- Olson KR, Dombkowski RA, Russell MJ, Doellman MM, Head SK, Whitfield NL, et al. Hydrogen sulfide as an oxygen sensor/transducer in vertebrate hypoxic vasoconstriction and hypoxic vasodilation. J Exp Biol 2006;209:4011-23.
- Zhang QY, Du JB, Zhou WJ, Yan H, Tang CS, Zhang CY. Impact of hydrogen sulfide on carbon monoxide/heme oxygenase pathway in the pathogenesis of hypoxic pulmonary hypertension. Biochem Biophys Res Commun 2004;317:30-7.
- Jin HF, Du JB, Li XH, Wang YF, Liang YF, Tang CS. Interaction between hydrogen sulfide/cystathionine gamma-lyase and carbon monoxide/heme oxygenase pathways in aortic smooth muscle cells. Acta Pharmacol Sin 2006;27:1561-6.
- Yan H, Du JB, Tang CS, Geng B, Jiang HF. Changes in arterial hydrogen sulfide (H2S) content during septic shock and endotoxin shock in rats. J Infect 2003;47:155-60.
- Wang J, Hegele RA. Genomic basis of cystathioninuria (MIM 219500) revealed by multiple mutations in cystathionine gamma-lyase (CTH). Hum Gene 2003;112:404-8.
- Geng B, Yang J, Qi Y, Zhao J, Pang Y, Du JB, et al. H2S generated by heart in rat and its effects on cardiac function. Biochem Biophys Res Commun 2004;313:362-8.
- O’Rourke B. Pathophysiological and protective roles of mitochondrial ion channels. J Physiol 2000;529:23-36.
- Gross GJ, Fryer RM. Sarcolemmal versus mitochondrial ATP-sensitive K+ channels and myocardial preconditioning. Circ Res 1999;84:973-9.
- Pan TT, Feng ZN, Lee SW, Moore PK, Bian JS. Endogenous hydrogen sulfide contributes to the cardioprotection by metabolic inhibition preconditioning in the rat ventricular myocytes. J Mol Cell Cardiol 2006;40:119-30.
- Zhu YZ, Wang ZJ, Ho P, Loke YY, Zhu YC, Huang XW, et al. Hydrogen sulfide and its cardioprotective effects in myocardial ischemia in experimental rats. J Appl Physiol 2006;102:261-8.
- Geng B, Chang L, Pan C, Qi Y, Zhao J, Pang Y, et al. Endogenous hydrogen sulfide regulation of myocardial injury induced by isoproterenol. Biochem Biophys Res Commun 2004;318:756-63.
- Patacchini R, Santicioli P, Giuliani S, Maggi CA. Pharmacological investigation of hydrogen sulfide (H2S) contractile activity in rat detrusor muscle. Eur J Pharmacol 2005;509:171-7.
- Maggi CA, Santicioli P, Giuliani S, Furio M, Meli A. The capsaicin-sensitive innervation of the rat urinary bladder: further studies on mechanisms regulating micturition threshold. J Urol 1986;136:696-700.
- Birder LA, Apodaca G, De Groat WC, Kanai AJ. Adrenergic- and capsaicin-evoked nitric oxide release from urothelium and afferent nerves in urinary bladder. Am J Physiol 1998;275:226-9.
- Maggi CA, Patacchini R, Giachetti A, Meli A. Tachykinin receptors in the circular muscle of the guinea-pig ileum. Br J Pharmacol 1997;101:996-1000.
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 1997;389:816-24.
- Tominaga M, Numazaki M, Iida T, Moriyama T, Togashi K, Higashi T, et al. Regulation mechanisms of vanilloid receptors. Novartis Found Symp 2004;261:4-12.
- Zygmunt PM, Julius I, Di Marzo I, Hogestatt ED. Anandamide–the other side of the coin. Trends Pharmacol Sci 2000;21:43-4.
- Hwang SW, Cho H, Kwak J, Lee SY, Kang CJ, Jung J, et al. Direct activation of capsaicin receptors by products of lipoxygenases: endogenous capsaicin-like substances. Proc Natl Acad Sci USA 2000;97:6155-60.
- Huang SM, Bisogno T, Trevisani M, Al-Hayani A, De Petrocellis L, Fezza F, et al. An endogenous capsaicin-like substance with high potency at recombinant and native vanilloid VR1 receptors. Proc Natl Acad Sci USA 2002;99:8400-5.
- Wimalawansa SJ. Antihypertensive effects of oral calcium supplementation may be mediated through the potent vasodilator CGRP. Am J Hypertens 1993;6:996-1002.
- Birder LA, Nakamura Y, Kiss S, Nealen ML, Barrick S, Kanai AJ, et al. Altered urinary bladder function in mice lacking the vanilloid receptor TRPV1. Nat Neurosci 2002;5:856-60.
- Inoue K, Koizumi S, Fuziwara S, Denda S, Inoue K, Denda M. Functional vanilloid receptors in cultured normal human epidermal keratinocytes. Biochem Biophys Res Commun 2002;291:124-9.
- Xin H, Tanaka H, Yamaguchi M, Takemori S, Nakamura A, Kohama K. Vanilloid receptor expressed in the sarcoplasmic reticulum of rat skeletal muscle. Biochem Biophys Res Commun 2005;332:756-62.
- Trevisani M, Smart D, Gunthorpe MJ, Tognetto M, Barbieri M, Campi B, et al. Ethanol elicits and potentiates nociceptor responses via the vanilloid receptor-1. Nat Neurosci 2002;5:546-51.
- Teague B, Asiedu S, Moore PK. The smooth muscle relaxant effect of hydrogen sulphide in vitro: evidence for a physiological role to control intestinal contractility. Br J Pharmacol 2002;137:139-45.
- Li L, Bhatia M, Zhu YZ, Zhu YC, Ramnath RD, Wang ZJ, et al. Hydrogen sulfide is a novel mediator of lipopolysaccharide-induced inflammation in the mouse. FASEB J 2005;19:1196-8.