
Display of aggregation-prone ligand binding domain of human PPAR gamma on surface of bacteriophage lambda1
Introduction
Nuclear receptors (NR) are a superfamily of ligand-activated transcriptional factors that are involved in diverse physiological functions[1,2]. Nuclear receptors share a common protein structure, including a highly conserved DNA-binding domain (DBD) responsible for binding to their corresponding hormone response elements located in the promoter region of their target genes, and a less-conserved ligand-binding domain (LBD) responsible for hormone binding, dimerization, and ligand-dependent activation. The nuclear receptors are activated by ligands binding to the hydrophobic ligand binding pockets in LBD, which triggers a conformational change in the receptor proteins. Because their activity can be modulated by small molecules that can be easily modified, nuclear receptors have become promising pharmacological targets for drug development[3].
Numerous techniques and tools for the screening of small molecular ligands have emerged over the past decade, but a major challenge for traditional ligand screening methods has been to express the protein of interest in soluble form and purify it efficiently. Mass production and purification of well-expressed and highly soluble proteins for traditional screening is a major obstacle, because high-level expression of recombinant proteins in E coli will always result in the formation of insoluble inclusion bodies. Thus, there is a significant need to develop a rapid protein expression and purification approach for high-throughput screening.
Phage display is a method for the expression of peptides, proteins or antibody fragments fused to the surface of phage particles. The methodology combines the protein expression and purification process with a subsequent rapid selection procedure[4,5]. Therefore, it is a potential tool for the production of proteins that could be used in the screening of ligands. Lytic bacteriophages such as lambda, T4 or T7 have been found to be useful for displaying foreign proteins[6–8]. Using the lambda capsid protein pD appears to be a particularly attractive option, because a variety of large proteins or protein domains, such as β-galactosidase, β-lactamase, and recombinant proteins encoded by cDNA have been successfully displayed on the surface of lambda phage as fusions to its N or C-terminus[9–12]. However, these proteins are exclusively soluble when expressed in bacterial systems, so the potential for lambda phage to display proteins that can be aggregated, such as nuclear receptors, is still unknown.
The peroxisome proliferator-activated receptors (PPAR) are members of the nuclear receptor superfamily, and are important in regulating lipid and glucose homeostasis[3,13]. One isoform, PPARγ, plays an important role in adipocyte differentiation and lipid homeostasis, and is a drug target for a variety of diseases, including obesity, diabetes, atherosclerosis and cancer[3]. However, the existing PPARγ ligands on the market have been associated with hepatotoxicity, which has resulted in the withdrawal of some of the PPAR ligands[14]. Therefore, developing a superior PPARγ LBD model would be helpful in the search for more effective and safe PPARγ ligands that have the potential to treat human diseases involving glucose and/or lipid disruption.
Similar to the other members in the nuclear receptor superfamily, the production and subsequent purification of large amounts of soluble PPARγ protein are difficult because of the hydrophobic nature of the ligand-binding pocket in the LBD. Because pD, a protein of the lambda capsid, has been described to have chaperone properties that can increase the expression level of soluble heterologous proteins in the cytoplasm of E coli[15], it could be used to express and incorporate the pD-LBD fusion protein on the surface of bacteriophage lambda.
In order to develop and implement phage surface display technology for the ligand-binding domain of NR, the PPARγ LBD was expressed as fusion protein of LBD-g3p and pD-LBD in E coli cells. The solubility characteristics of these two systems were compared to determine the phage display system most appropriate for PPARγ LBD expression. Finally, the PPARγ LBD fused to the appropriate capsid protein was characterized by Western blotting and phage capture assays.
Materials and methods
Plasmid construction A polymerase chain reaction (PCR) fragment of the human PPARγ2 LBD (amino acids 201–505, GenBank accession N
The p171-LBD expression vector was constructed by inserting a PCR fragment of human PPARγ2 LBD, which was amplified by the primers PP_Fwd2 (5'-CG
Protein expression and purification E coli strain BB4 was transformed with pCGMT-LBD and p171-LBD and grown to an OD600 of 0.6 in 50 mL LB media containing 1% (w/v) glucose and 60 mg/L ampicillin at 37 °C. Afterwards, the cells were induced with 1 mmol/L isopropyl-D-thiogalacto-pyranoside (IPTG) for an additional 6 h at 30 °C, then the cells were collected by centrifugation. After being washed three times with sonication buffer [50 mmol/L Tris, pH 8.0, 0.15 mol/L NaCl, 1 mmol/L ethylenediamine tetraacetic acid (EDTA)], the pellets were resuspended in 50 mL sonication buffer again and half of them were disrupted by sonication at 4 °C. After the lysate was centrifuged at 12000×g for 15 min, the resultant supernatant was recovered and the resultant precipitate that had the insoluble fraction was resuspended in 25 mL sonication buffer.
Polyclonal antibody preparation E coli strain BL21(DE3) was transformed with pET-hPPGLBD, then grown to an OD600 of 0.6 in LB media containing 60 mg/L ampicillin at 37 °C, then the cells were induced with 1 mmol/L IPTG for another 6 h at 30 °C. The cells were harvested and disrupted by sonication as described in the previous section. The human PPARγ2 LBD was expressed as inclusion bodies, whose homogeneity was estimated to be greater than 90% by visual inspection of Coomassie brilliant blue-stained sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels. The isolated inclusion bodies were used as antigens to immunize mice.
The p171Bio3 vector containing lambda capsid gene gpD was transformed into E coli BB4, which were then grown to an OD600 of 0.6 in LB media and induced with 1 mmol/L IPTG for 6 h at 30 °C. The cell extracts from sonication were applied to a 12% SDS-polyacrylamide gel. After electro-phoresis, the gels were soaked in 100 mL 0.3 mol/L KCl at 4 °C for 15 min. When the pD protein band in the gel became white, the band was cut out, frozen and thawed. Proteins in it were recovered with 1 mL 0.1 mol/L phosphate-buffered saline (PBS) and used as antigens for immunization.
Polyclonal antibodies to human PPARγ LBD (anti-LBD) or lambda pD (anti-pD) were prepared by immunizing female BALB/c mice with 50 µg of the recombinant protein emulsified with an equal volume of Freund’s complete adjuvant. The mice were given booster immunizations three times every 10 d with the same amount of antigen in Freund’s incomplete adjuvant. Ten days after the last immunization, blood was collected for testing of antibody reactivity. Afterwards, sera from the mice that contained the polyclonal antibodies were collected.
Lambda lysogen preparation and lambda phage rescue and titration Lysogenic BB4 was generated by infecting E coli strain BB4 with λDam15 b538 cIts857 nin5 Sam100, then selecting lambda lysogens at 32 °C. The prophage contained 78.5% of the genome of the wild type phage and an amber mutation in the gpD gene[18].
For lambda phage rescue, the lysogenic BB4 strain transformed with p171Bio3 or p171-LBD was grown under non-inducing conditions (below 38 °C) to an OD600 of 0.3 at 32 °C in 50 mL LB media containing 0.2% maltose, 0.1% glucose and 10 mmol/L MgSO4 with agitation, then induced at 42 °C for 15 min to inactivate the temperature-sensitive cIts857 repressor. IPTG was then added to the culture to a concentration of 1 mmol/L and incubated at 38 °C for an additional 3 h with vigorous agitation. After 1 mL chloroform was added to the culture to complete cell lysis, the culture was incubated in a shaker for an additional 15 min. The released phage particles in the culture were purified by two rounds of standard PEG and NaCl precipitation, and the resultant phage pellets were resuspended in 5 mL SM buffer (0.1 mol/L NaCl, 10 mmol/L MgSO4, 50 mmol/L Tris, 0.01% gelatin, pH 7.5), and stored at 4 °C.
For lambda phage titer determination, lambda phage samples were serially diluted in SM buffer, then mixed with 0.2 mL fresh cultured BB4 bacteria (OD600 of 0.5) grown in LB medium containing 0.2% (w/v) maltose and 10 mmol/L MgSO4. After 30 min of incubation at 37 °C, infected cells were mixed with 3 mL of molten LB top agar containing 0.2% maltose, 10 mmol/L MgSO4 and poured immediately onto LB plates, which were then incubated overnight at 37 °C. Phage plaque number on the plate was counted and the titer of lambda phage was calculated.
SDS-PAGE and Western blot analysis The protein expression of pCGMT, pCGMT-LBD, p171Bio3 and p171-LBD in the transformed BB4 strain was assayed. After cell lysis by sonication, samples of the supernatant fraction and the precipitated fraction were analyzed on a 10% SDS-PAGE gel. For the expression comparison of PPARγ LBD fused to g3p or pD, equal amounts of total cell protein, supernatant fraction and precipitated fraction after sonication were analyzed by using standard SDS-PAGE. The bands were visualized by using Coomassie brilliant blue staining and Western blotting with anti-LBD polyclonal antibody. For phage electro-phoresis, 1×109 lambda phage particles were mixed with the loading buffer and boiled for 15 min, then applied to a 10% SDS-PAGE gel and analyzed with anti-LBD or anti-pD polyclonal antibody.
For Western blot analysis, separated proteins in the gels were electrophoretically transferred onto a PVDF membrane (Immobilon-P, Millipore) at 400 mA for 90 min. The membrane was blocked in a blocking buffer [3% bovine serum albumin (BSA) in Tris-buffered saline (TBS), 150 mmol/L NaCl, pH 7.4] for 2 h at room temperature, and then incubated with either a primary antibody (anti-LBD or anti-pD polyclonal antibody, diluted 1:1000 in the blocking buffer) at 37 °C for 1 h. After three washes in TBST (0.1% Tween-20 in TBS buffer, pH 7.4, 10 min for each wash), the blots were incubated with a horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG secondary antibody (Calbiochem; at a dilution of 1:1000 in the blocking buffer) at room temperature for 1 h. The blots were then washed in TBST (three times, 10 min for each wash), then stained with HRP substrate diaminobenzidine (DAB).
Phage capture assay The plates were coated with the serum containing anti-LBD polyclonal antibody in carbonate buffer (50 mmol/L NaHCO3, pH 9.6) overnight at 4 °C (100 µL/well, n=6), and the control wells were coated with the serum from the non-immunized mice (n=3). After discarding the coating solution, each well was incubated with 200 µL blocking solution (2% BSA in PBS, 0.05% Tween-20) for 2 h at 37 °C. A total of 1×108 λp171Bio3 particles per well were added to three wells coated with anti-LBD antibody, and the same amount of λp171-LBD was added to the other three wells coated with anti-LBD antibody and the three wells coated with control serum, and incubated for 1 h at 37 °C with gentle agitation. Afterwards, the plate was washed three times with 200 µL washing solution (PBS, 0.05% Tween-20, 10 mmol/L MgSO4) and once with PBS (10 mmol/L MgSO4). The lambda phages binding to each well were recovered by directly adding 200 µL of fresh cultured BB4 cells. After 30 min of incubation at 37 °C, the phage titer of the mixture was determined as described earlier. The statistical significance of the differences between the captured phage titers was assessed by using the paired Student’s t-test, and the level of statistical significance was set at P<0.05.
Results
Construction of p171-LBD, pCGMT-LBD and pET-hPPGLBD expression vectors Plasmid p171-LBD (Figure 1) was constructed by inserting the PCR-amplified fragment of the PPARγ2 gene (coding amino acids 201–505) into the SpeI-NotI site of p171Bio3, which contained the strong tac promoter and the lacIq gene, and thus could tightly control the expression of the downstream lambda capsid gene gpD. PPARγ LBD was expressed as a fusion to the carboxyl terminus of pD (approximately 11 kDa), which is one of the lambda phage head proteins that form the protruding trimeric structure essential for the stability of the capsid, and is also used as a carrier protein for lambda phage display[6,11].
The pCGMT-LBD plasmid (Figure 1) was constructed similarly, except that different cloning sites were used. The coding region of PPARγ LBD was cloned into the 5' terminus of g3 in phagemid pCGMT, which contained a lac-promoter, a pelB signal sequence, and an amber codon between the fusion protein and the truncated g3p. PPARγ LBD would be expressed as a fusion protein in an amber suppressor strain, such as BB4, to the N terminus of g3p, which is a minor coat protein of phage M13[19], and the most commonly used carrier protein for displaying large proteins in a filamentous phage display system because it is less sensitive to the size of protein inserts.
Plasmids of pET-hPPGLBD were constructed by cloning the PCR fragment of the PPARγ2 LBD into BamHI and SalI sites in pET-21a, in an attempt to express a large amount of PPARγ LBD protein in E coli in order to obtain antigens for generating mouse anti-LBD antibodies.
Lambda capsid protein pD was a more suitable carrier protein for displaying PPARγ LBD than g3p In order to assess which of the 2 phage display systems (ie, the filamentous phage system or the lytic lambda display system) was more appropriate for displaying PPARγ LBD, a comparison between the expression of PPARγ LBD fused to capsid protein g3p or pD was firstly performed.
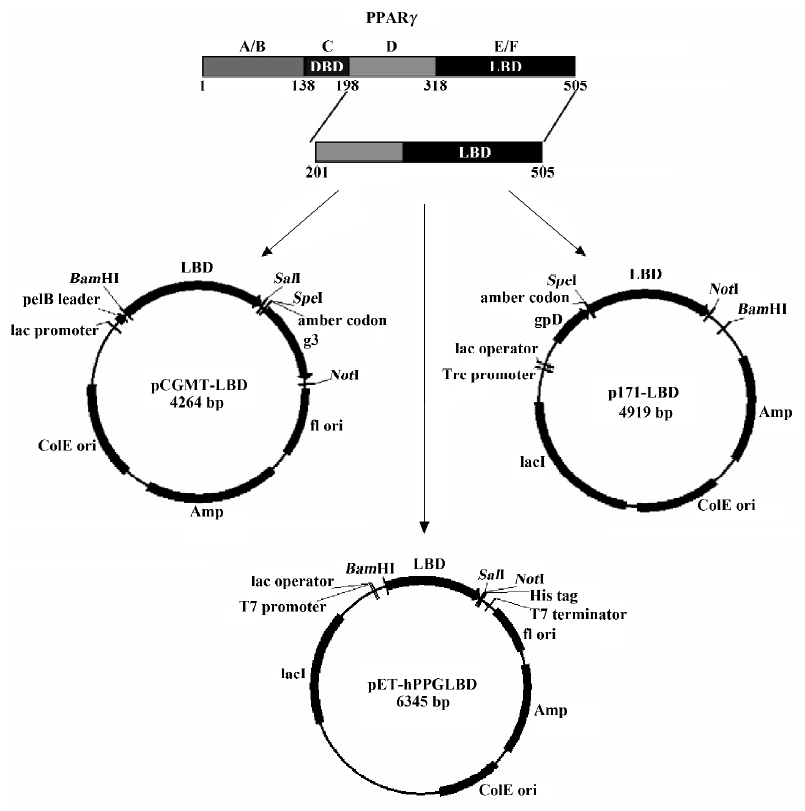
pCGMT-LBD and p171-LBD produced the LBD-g3p and pD-LBD fusion proteins, respectively, when expressed in the amber repressor bacterial strain BB4 (supE). The estimated molecular weights of these 2 proteins were 55 kDa (the carboxyl-terminal PPARγ protein is approximately 35 kDa, whereas the truncated g3p accounts for approximately 20 kDa) and 46 kDa (pD is approximately 11 kDa), respectively. The proteins in the induced total cells, the supernatant and precipitated fractions of sonication lysate were analyzed with Coomassie brilliant blue staining (Figure 2A) or Western blotting with anti-LBD antibody (Figure 2B). A protein band with a molecular weight of 55 kDa, corresponding to LBD-g3p, was observed in the total cell lysate (lane 3), and a 35-kDa band corresponding to pD-LBD was observed as well (lane 6), indicating that PPARγ LBD could be expressed as a fusion protein after induction. However, almost all of the LBD-g3p protein was insoluble (lane 5), and no LBD-g3p protein was found in the supernatant fraction (on the basis of anti-LBD antibody detection; lane 4). Although over-expression of pD-LBD protein could make this fusion protein form insoluble inclusion bodies as well (lane 8), a reasonable amount was expressed in a soluble form (lane 7). Furthermore, by density analysis it was shown that the soluble protein accounted for approximately 40% of the total expressed LBD. These results show that the pD-LBD fusion protein was partially soluble when expressed in E coli, whereas the LBD-g3p fusion protein was detected only in the insoluble fraction. Because solubility was a prerequisite for displaying foreign proteins on the phage surface, the lambda capsid protein pD seems to be a more appropriate carrier protein for displaying PPARγ LBD. Thus PPARγ LBD expressed with the lambda system rather than the filamentous phage system was further characterized.
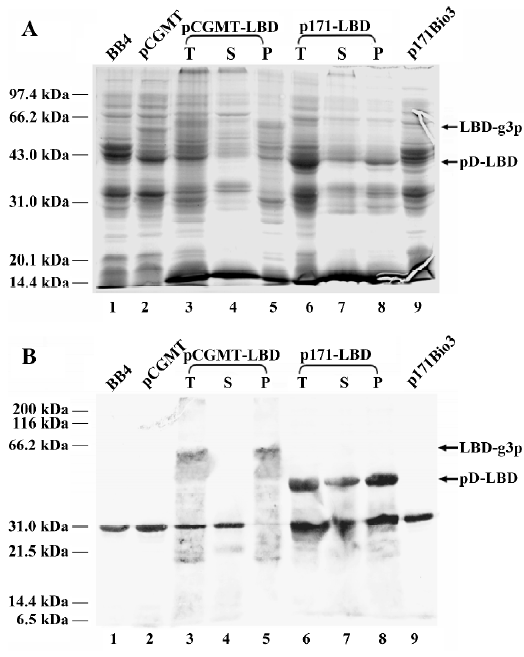
Part of PPARγ LBD fused to pD was expressed in soluble form in bacterial cytoplasm Additional studies for assessing the solubility of expressed pD-LBD under induction and noninduction conditions were performed. Crude protein extracts from BB4 cells transformed with vector p171-LBD or parent vector p171Bio3 were analyzed by SDS-PAGE (Figure 3A) and western blotting (Figure 3B). As shown in Figure 3B, a small amount of soluble pD-LBD (46 kDa) was detected in the supernatant (Figure 3B, lane 6, without IPTG induction), whereas no pD-LBD was detected in the precipitated fraction (Figure 3B, lane 7). After IPTG induction (Figure 3B, lanes 8 and 9), the expression of the pD-LBD fusion protein increased markedly. Because the pD proteins could only improve their solubility to some extent, most of the overexpressed fusion protein under induction conditions aggregated in an insoluble form (Figure 3B, lane 9), whereas soluble pD-LBD increased moderately with IPTG induction (Figure 3B, lane 8). The relative amount of pD-LBD fusion protein in the supernatant and precipitated fraction was estimated from the PVDF membrane by densitometry measurements, which indicated that approximately 30% of total PPARγ LBD fusion proteins were expressed in soluble form. Although a reasonable number of them were expressed as insoluble inclusion bodies, quite a few of the hydrophobic PPARγ LBD could maintain their solubility after being fused to the lambda pD protein, which might be sufficient for the protein to be displayed on the surface of lambda phage.
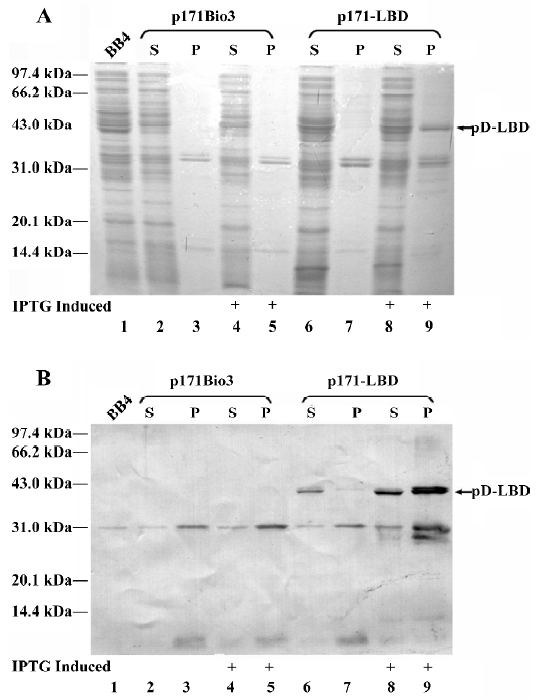
PPARγ LBD could be incorporated into lambda phage capsid Protein extracts from rescued lambda phage particles of λp171Bio3 or λp171-LBD were probed with anti-LBD (Figure 4A) or anti-pD (Figure 4B) polyclonal antibody, respectively, after electrophoresis on a 10% SDS-PAGE gel, and were transferred onto a PVDF membrane.
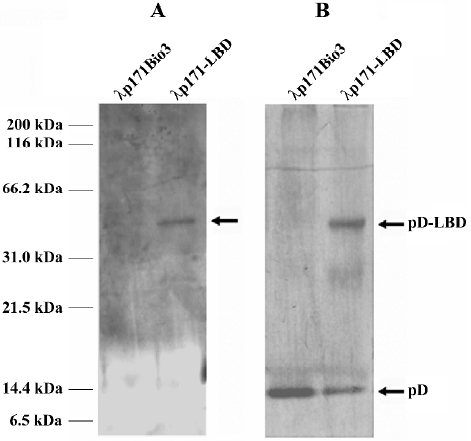
Western blotting analysis with an antibody against PPARγ LBD produced a prominent band of 46 kDa in the λp171-LBD lane, whereas no corresponding band was detected in the control lane of λp171Bio3 (Figure 4A). More-over, the result was validated by probing with anti-pD polyclonal antibody. Two bands, with molecular weights of approximately 11 kDa and 46 kDa, were detected (Figure 4B), which represented protein pD from the gpD gene of the integrated prophage genome, and the pD-LBD fusion protein from the gpD gene of the plasmid, respectively. The ratio of the proteins in the two bands represented the level of the pD-LBD fusion protein incorporated into lambda phage. Density comparisons of the two bands of λp171-LBD in Figure 4B indicated that the amount of fusion pD accounted for nearly 28% of the total pD protein content on lambda phage. Because there were 405 copies of protein pD on the capsid of wild-type lambda, we could estimate that the average number of PPARγ LBD incorporated into the lambda phage capsid was approximately 115 per phage particle. In summary, we conclude that the ligand-binding domain of PPARγ could be efficiently incorporated into lambda phage particles.
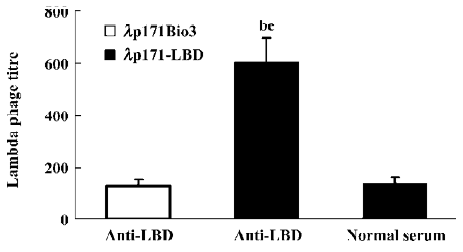
Phage capture assay indicated that PPARγ LBD was expressed on the surface of lambda phage To identify the sites displaying PPARγ LBD on lambda phage, a phage capture assay was performed. The titer of phage λp171-LBD was compared to that of λp171Bio3 after binding to wells coated with the anti-LBD antibody. From the results presented in Figure 5, the titer of captured λp171-LBD phage with incorporation of PPARγ LBD was approximately four times more than that of captured λp171Bio3 (P<0.01), indicating that PPARγ LBD incorporated in phage capsids is selectively recognized by mouse antibodies. However, on the wells coated with mouse normal serum rather than anti-LBD antibody serum, the titer of λp171-LBD captured decreased markedly (P<0.01), which indicated that only anti-LBD antibody could capture λp171-LBD. It was clear that the PPARγ LBD incorporated in λp171-LBD phage exhibited specific binding to the immobilized anti-LBD antibody. In summary, we conclude from our results that the PPARγ LBD was displayed on the surface of the bacteriophage lambda capsid.
Discussion
Nuclear receptors are a large family of transcription factors involved in many important metabolic processes. To date, 48 members have been identified in the human genome, and all the members of this family have a modular structure composed of six domains (A−F)[2]. Endogenous ligands have not been identified for all NR. NR are termed “orphan receptors” if their endogenous ligands have not yet been discovered, and “adopted” when their endogenous ligands are identified. The identification of new ligands for NR not only provides the opportunity to elucidate their function, but can also bring about the discovery of potential therapeutic agents for human diseases[3,20].
Many in vitro high-throughput screening methods have been applied in an effort to search for novel ligands for these nuclear receptors; however, the production of large quantities of highly soluble proteins as well as the subsequent purification of these proteins is the main obstacle to overcome in these high-throughput assays for novel ligands using traditional screening systems. E coli cells offer a convenient and inexpensive expression system for the production of human proteins; however, the high-level expression of recombinant proteins in E coli often results in the formation of insoluble inclusion bodies. The commonly used approaches to address the solubility problems of recombinant proteins have focused on optimizing expression conditions or on fusion of protein partners, such as glutathione-S-transferase (GST)[21], maltose binding protein (MBP)[22] and thioredoxin (Trx)[23]. However, these methods are not always effective, especially for very hydrophobic proteins.
Phage display techniques can couple protein expression and purification with the subsequent screening steps after the protein is assembled on the phage surface, which can circumvent the problems associated with protein purification in conventional affinity screening methods. By affinity binding with a given target, proteins can be isolated and identified without consideration of protein purification and yield, and vice versa. Previous reports have demonstrated that large proteins can be displayed on the phage surface, and this technique has been proven to be useful in high-throughput screening for antagonists of the receptor and other proteins[5]. However, these previous successfully displayed proteins share the properties of soluble proteins when expressed in E coli, such as antibody fragments (scFv), enzymes[11,12], bacterial proteins (staphylococcal protein A)[24] and virus capsid proteins (HIV-1 p24, and HCV)[9]. Whether aggregation-prone proteins can be displayed on the phage surface was not elucidated.
The four types of variant PPARγ, which come from alternative promoters and differential splicing[4], have the same LBD and C-terminus. Because its LBD and DBD function independently, as in the other nuclear receptors, it is possible to express a truncated PPARγ or an isolated PPARγ domain to study its functions and binding characteristics. However, the ligand-binding pocket in the LBD makes expression more difficult because of its hydrophobic nature[25]. Our experiments indicated that nearly all the recombinant PPARγ LBD is in an insoluble form when expressed in a pET system for antigen preparation (unpublished data).
Other reports have also indicated that the ability to remain soluble is a prerequisite for a protein to be incorporated into the phage surface. An improvement in protein solubility after partner fusion or molecular chaperone coexpression can increase the incorporation efficiency of fusion proteins displayed on the phage surface[26,27]. However, unlike the traditional affinity screening methods, which require the purification of large quantities of soluble protein, phage display only requires the protein to be moderately soluble, and a small amount of soluble protein is sufficient to be expressed on the phage surface. Furthermore, lytic bacteriophages such as lambda, T4 or T7 have been shown to be promising systems for the of display foreign proteins, because the encapsidation of the foreign fusion protein is an intracellular event, thus making the secretion of the fusion protein a less-demanding process and gaining an advantage over the filamentous phage for displaying foreign proteins[6–8]. Our results showed that protein pD improved the solubility of pD-LBD to some extent, and that the fusion protein was soluble enough for phage display of PPARγ LBD. This might be a useful approach for circumventing the expression and purification problems in traditional screening methods. Moreover, proteins as large as β-galactosidase have been successfully displayed on lambda phage surfaces as fusions to the amino or carboxyl terminus of protein pD[11,12], which implies that the size limit for proteins in lambda phage display systems is not very strict. Thus the pD lambda phage system is superior to other phage display systems, and is particularly appropriate for the expression of large proteins that tend to form insoluble inclusion bodies.
The lambda display system based on protein pD is a polyvalent display system, which is useful for the efficient selection of ligands with either low or moderate binding affinity. Previous studies indicate that the percentage of fusion protein incorporated in the capsid can reach up to 90% of the total pD protein content[24,28], which makes it difficult to select high-affinity binding ligands. However, the efficiency of selection of high-affinity ligands can be improved, because the expression of pD fusion protein can be regulated by the promoter. The pD-LBD fusion protein displayed on the lambda phage capsid represented nearly one-third of the total pD protein content. By altering the ratio of wild type pD to pD fusion protein, it is possible to change the valency of fusion protein on the lambda surface.
Our results demonstrated that PPARγ LBD fusion protein was incorporated into the lambda phage capsid and expressed on the surface of the lambda phage. Further studies remain to be conducted to characterize the activity of the displayed PPARγ LBD, including binding assays with known PPARγ ligands, as well as pilot assays involving ligand screening by directly panning phage-displayed fusion proteins against immobilized molecules, or small molecule competition binding between test compounds and known complexes of phage-displayed PPARγ LBD and its natural ligand. This system may be a new alternative for expressing foreign proteins that tend to be insoluble when using conventional approaches, and quite possibly has great value for downstream screening of novel PPARγ ligands.
Acknowledgement
We gratefully acknowledge the gift of the E coli strain and plasmid from Dr Alfredo NICOSIA (IRBM, Rome, Italy) and Dr Hitoshi NISHIZAWA (Osaka University, Osaka, Japan).
References
- Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, et al. The nuclear receptor superfamily: the second decade. Cell 1995;83:835-9.
- Weatherman RV, Fletterick RJ, Scanlan TS. Nuclear-receptor ligands and ligand-binding domains. Annu Rev Biochem 1999;68:559-81.
- Berkenstam A, Gustafsson JA. Nuclear receptors and their relevance to diseases related to lipid metabolism. Curr Opin Pharmacol 2005;5:171-6.
- Li M. Applications of display technology in protein analysis. Nat Biotechnol 2000;18:1251-6.
- Smith GP, Petrenko VA. Phage display. Chem Rev 1997;97:391-410.
- Hoess RH. Bacteriophage lambda as a vehicle for peptide and protein display. Curr Pharm Biotechnol 2002;3:23-8.
- Ren Z, Black LW. Phage T4 SOC and HOC display of biologically active, full-length proteins on the viral capsid. Gene 1998;215:439-44.
- Danner S, Belasco JG. T7 phage display: a novel genetic selection system for cloning RNA-binding proteins from cDNA libraries. Proc Natl Acad Sci USA 2001;98:12954-9.
- Santini C, Brennan D, Mennuni C, Hoess RH, Nicosia A, Cortese R, et al. Efficient display of an HCV cDNA expression library as C-terminal fusion to the capsid protein D of bacteriophage lambda. J Mol Biol 1998;282:125-35.
- Santi E, Capone S, Mennuni C, Lahm A, Tramontano A, Luzzago A, et al. Bacteriophage lambda display of complex cDNA libraries: a new approach to functional genomics. J Mol Biol 2000;296:497-508.
- Sternberg N, Hoess RH. Display of peptides and proteins on the surface of bacteriophage lambda. Proc Natl Acad Sci USA 1995;92:1609-13.
- Mikawa YG, Maruyama IN, Brenner S. Surface display of proteins on bacteriophage lambda heads. J Mol Biol 1996;262:21-30.
- Schoonjans K, Staels B, Auwerx J. The peroxisome proliferator activated receptors (PPARS) and their effects on lipid metabolism and adipocyte differentiation. Biochim Biophys Acta 1996;1302:93-109.
- Watkins PB, Whitcomb RW. Hepatic dysfunction associated with troglitazone. N Engl J Med 1998;338:916-7.
- Forrer P, Jaussi R. High-level expression of soluble heterologous proteins in the cytoplasm of Escherichia coli by fusion to the bacteriophage lambda head protein D. Gene 1998;224:45-52.
- Nishizawa H, Yamagata K, Shimomura I, Takahashi M, Kuriyama H, Kishida K, et al. Small heterodimer partner, an orphan nuclear receptor, augments peroxisome proliferator-activated receptor gamma transactivation. J Biol Chem 2002;277:1586-92.
- Gao C, Lin CH, Lo CL, Mao S, Wirsching P, Lerner RA, et al. Making chemistry selectable by linking it to infectivity. Proc Natl Acad Sci USA 1997;94:11777-82.
- Enquist L, Sternberg N. In vitro packaging of lambda Dam vectors and their use in cloning DNA fragments. Methods Enzymol 1979;68:281-98.
- Parmley SF, Smith GP. Antibody-selectable filamentous fd phage vectors: affinity purification of target genes. Gene 1988;73:305-18.
- Gronemeyer H, Gustafsson JA, Laudet V. Principles for modulation of the nuclear receptor superfamily. Nat Rev Drug Discov 2004;3:950-64.
- Smith DB, Johnson KS. Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene 1988;67:31-40.
- di Guan C, Li P, Riggs PD, Inouye H. Vectors that facilitate the expression and purification of foreign peptides in Escherichia coli by fusion to maltose-binding protein. Gene 1988;67:21-30.
- LaVallie ER, DiBlasio EA, Kovacic S, Grant KL, Schendel PF, McCoy JM. A thioredoxin gene fusion expression system that circumvents inclusion body formation in the E coli cytoplasm. Biotechnology (NY) 1993;11:187-93.
- Gupta A, Onda M, Pastan I, Adhya S, Chaudhary VK. High-density functional display of proteins on bacteriophage lambda. J Mol Biol 2003;334:241-54.
- Mi LZ, Rastinejad F. Large scale production of nuclear receptor ligand-binding domains. In: Lieberman BA, editor. Steroid receptor methods: protocols and assays. Totowa, NJ: Humana Press; 2001. p 81−90.
- Bothmann H, Pluckthun A. Selection for a periplasmic factor improving phage display and functional periplasmic expression. Nat Biotechnol 1998;16:376-80.
- Hayhurst A, Harris WJ. Escherichia coli skp chaperone coexpression improves solubility and phage display of single-chain antibody fragments. Protein Expr Purif 1999;15:336-43.
- Zucconi A, Dente L, Santonico E, Castagnoli L, Cesareni G. Selection of ligands by panning of domain libraries displayed on phage lambda reveals new potential partners of synaptojanin 1. J Mol Biol 2001;307:1329-39.