
Toosendanin interferes with pore formation of botulinum toxin type A in PC12 cell membrane1
Introduction
Botulinum neurotoxin (BoNT), an exotoxin produced by the bacterium Clostridium botulinum, is the etiological agent in botulism poisoning. Although there are 7 distinct serotypes (designated A–G), they have the same general molecular properties. Each serotype of BoNT consists of a ~100 kDa heavy chain (HC) and a ~50 kDa light chain (LC), linked by a single disulfide bond and non-covalent forces[1,2]. A 4-step mechanism consisting of binding, internalization, translocation and cleaving the soluble NSF accessory protein receptor (SNARE) protein is currently the accepted explanation for BoNT intoxication[3,4]. It is believed that BoNT enters cells via receptor-mediated endocytosis[4]. Exposure of the BoNT holotoxin to the acidic pH of the endosomes induces a conformational change of the disulfide-linked dichain toxin, allowing the LC protease to pass across the membrane through the putative HC channel and into the cytosol[5]. When the LC of the toxin reaches the cytosol, it acts as a zinc-dependent endoprotease to cleave polypeptides that are essential for exocytosis, which results in synaptic transmission blockade. It has been known for some time that BoNT is capable of forming ion channels in artificial bilayers[6–9] and PC12 cell membrane[10]. A recent study demonstrated that toxin translocation and channel formation were correlated[11]. The finding suggested to us that the BoNT channel represented a potential target for intervention to attenuate BoNT neurotoxicity.
Toosendanin (TSN, C30H38O11, FW=574), a triterpenoid derivative[12,13], is extracted from the bark of Melia toosendan Sieb et Zucc. It is used in Chinese traditional medicine as an ascarifuge[14]. Despite having some actions in common with BoNT, TSN has been shown to have a markedly antibotu-lismic effect both in vivo and in vitro. For example, TSN can prevent death in animals (mice, rats and monkeys) treated with several times the normal lethal dose of BoNT[15,16]. The time to paralysis of the neuromuscular junction after treatment with BoNT is lengthened by several times when the preparations were preincubated with TSN[16,17]. Our recent study showed that TSN treatment made synaptosomes resistant to BoNT/A-mediated proteolytic cleavage on their SNAP-25[18]. The protective effect did not result from inhibiting the endopeptidase activity of the toxin, but from interference with the approach of the LC to the substrate.
The purpose of this study was to investigate whether TSN affects BoNT/A-induced channels, and hence protects SNAP-25 from cleavage. NGF-differentiated PC12 cells were used in this study. The cells become very sensitive to BoNT after differentiation[19].
Materials and methods
Cell culture PC12 cells were obtained from the Type Culture Collection of the Chinese Academy of Sciences and were serially passaged in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, NY, USA) supplemented with 5% fetal calf serum (HyClone, UT, USA). The cells were plated at low density in culture medium supplemented with 100 ng/mL nerve growth factor (NGF, 2.5S) in 35-mm plastic dishes (Corning, NY, USA). After a minimum of 4 d, the cells were used for electrophysiological recordings.
Solutions and chemicals The standard bath solution used for the inside-out patch recording contained 200 mmol/L CsCl and 1 mmol/L dithiothreitol, and was buffered to pH 7.0 with 5 mmol/L Na·MOPS (3-(N-morpholino)propane-sulfonate). The solution to fill the pipettes contained 200 mmol/L CsCl and was buffered to pH 5.3 with 5 mmol/L Na·MES (2-(N-morpholino)ethanesulfonate). All solutions were filtered at 0.45 μm before use. Unless otherwise specified, all chemicals were from Sigma (St Louis, MO, USA). The 500 kDa BoNT/A was from Wako (Japan). The toosendanin used in this work was a sample recrystallized in ethanol with a purity of >98%[20]. Toosendanin was added to the standard bath or pipette solution at a final concentration of 35 µmol/L. Previous studies have demonstrated that at a concentration of 17 µmol/L, TSN abolishes the action of BoNT/A to cleave its substrate[18]. Here, a higher concentration was used.
Electrophysiological recordings Patch pipettes were pulled from 1.5 mm (outer diameter) capillary glass (type 95, Shanghai Institute of Physiology, Shanghai, China) using a Narishige PP-83 electrode puller (Narishige, Japan). The tip of each pipette was coated with N-trimethylsilydiethylamine and fire-polished on a microforge (FP-1, Shanghai Institute for Biological Sciences, CAS). Tip resistance was 4–8 MΩ when filled with pipette solution. Single channel currents were recorded in the inside-out configuration[21] using a patch-clamp amplifier (Axopatch 200A, Axon Instruments, Foster City, CA, USA) at room temperature (22–25 °C). After the pipettes were removed from the cell somas, the isolated membranes were exposed to symmetrical salt solutions with a pH gradient that was acidic on the pipette, or extracellular, membrane surface. The previously intracellular side was exposed to a bath solution at neutral pH.
Data analysis Membrane current was filtered at 1 kHz and digitized at 20 kHz. Single channel analysis was performed using the pClamp 6.0.4 program (Axon Instruments). The membrane voltages are presented as the voltages at the pipette referenced to the grounded bath and represent the potentials on the side of the membrane where the toxin was applied. As a preliminary criterion for selecting BoNT-induced ion channels, membrane patches showing significant channel activity immediately following excision were discarded, because these were most likely endogenous ion channels. In fact, most of the patches of membrane were initially electrically silent. To generate toxin-induced ion channels in these patches, 20 µg/mL of 500 kDa BoNT/A was added to the pipette filling solution described earlier before formation of the patch. As an index of channel activity we used NPo; that is, the product of the number of channels present in the patch membrane (N) and the probability that a particular channel is open (Po).
All data are expressed as mean±SEM. Statistical analysis was performed with Student’s t-test, and P<0.05 was considered significant.
Results
BoNT/A formed ion channels in PC12 cell membrane No ion channel activity was detected in 8 patches examined in the absence of BoNT/A in the pipette over 20 min. With the toxin, spontaneous ion channel openings due to BoNT/A were observed in 27 of 31 patches examined after a delay of 2–9 min following first contact with the membrane patch (average time 3.5±1.9 min). Typical membrane ion channels formed by BoNT/A are shown in Figure 1A. Once these toxin-induced ion channels appeared they remained active until the gigohm patch seal decayed. During this period, the apparent conductance of individual channels increased with time after their first appearance. As shown in Figure 2A, for membrane patches exposed to BoNT/A, conductance through the toxin-induced channel became larger over time. The conductance of toxin-induced channels was essentially independent of membrane potential, as shown in the current-voltage plot in Figure 1D. In addition, channel activity and time-averaged current through the membrane patch increased with time (Figure 2B). In part, this represented an obvious increase in the number of individual active BoNT-induced channels in the membrane.

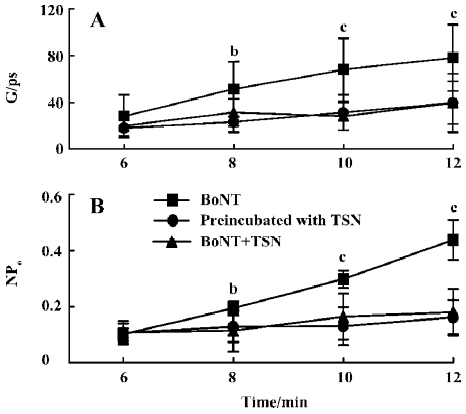
TSN interfered with the formation of the BoNT/A channels To test whether TSN affects the formation of the toxin-induced channel, two groups of experiments were carried out. In the first group of experiments, PC12 cells were preincubated with TSN for more than 30 min prior to the formation of the gigohm patch seal. In control experiments, no ion channel opening was observed without BoNT/A in the recording pipette (n=6). After BoNT/A was added into the pipette solution, spontaneous ion channel openings due to BoNT/A were observed in 16 of 36 patches examined after a delay of 8.0±2.5 min (Figure 1B). When these values are compared with those obtained without TSN, the probability of toxin-induced channel appearance was significantly decreased (P<0.05) and the delay before channel appearance was prolonged in the presence of TSN. In addition, the conductance of the toxin-induced channels observed under these conditions (Figure 1B, 2A) was smaller than that investigated without TSN. Under these conditions, 20 min after the channels appeared, the conductance was 56±9 pS (n=7), whereas without TSN the conductance was 120±11 pS (n=10). As seen in Figure 2B, after preincubation with TSN, the activities (NPo) of the channels that appeared were also reduced. Moreover, even in the membrane patches in which toxin-induced channels could form, not more than one active channel was observed in each patch examined. Under these conditions, the shape of the current-voltage curve was not changed (Figure 1D).
In the second group of experiments, TSN was added in the pipette solution in company with BoNT/A. In the control experiments with TSN only in the patch pipette, no ion channel opening was observed in 6 cells over 20 min. When BoNT/A was added accompanied by TSN, ion channel openings due to BoNT/A were observed in 14 of 31 patches examined after a delay of 5.5±1.2 min (Figure 1C). The channel formation rate was also substantially reduced. The conductance and activity of the toxin-induced channels under these conditions (Figure 2A, B) were similar to those observed in the first group of experiments, which were both smaller than those without TSN. These results indicate that TSN interfered with the pore-forming activity of BoNT/A.
TSN affected the BoNT/A-induced channels To investigate whether TSN blocks the toxin-induced channels, TSN was applied to the PC12 cells shortly after the ion channels appeared. As shown in Table 1, 10 min after perfusing the inside-out patch with the TSN-containing solution, neither the open probability (Po) nor the mean open time (mo) of the channels was affected (n=5). The conductance through the channels was significantly increased (P<0.05). However, the increased value was smaller than that obtained without TSN (Table 1). The results indicate that TSN delays the increase in the conductance of the BoNT/A-induced channels.

Full table
Discussion
Unlike many ligand or voltage-gated ion channels, the channels formed by BoNT are not easily identified by the action of well-identified inhibitors or agonists. BoNT-induced ion channels are usually identified by using the correlation between the presence or absence of the active toxin and the presence or absence of channel activity. In the present study, in the absence of BoNT/A, no channel activity was observed in 8 initially silent membrane patches examined. However, when the active toxin was present, ion channels formed in 27 of 31 patches with a delay similar to that found in a previous study[10]. The probability that the channels are not due to BoNT/A is very low. The properties of the channels we observed were similar to those previously reported[10]. The pH conditions in the present study, low pH on the side of toxin application and high pH on the membrane surface away from the toxin, were selected to optimize the formation of ion channels. These have the same orientation and magnitude of the pH gradients in endocytotic vesicles, which are thought to play a role in the translocation of toxin from an extracellular compartment to an intracellular site of action[22,23].
It is known that pore formation is involved in BoNT translocation[11]. In the present study we observed that TSN interfered with the formation of BoNT/A-induced ion channels. After the cells were preincubated with TSN, ion channels were observed in only 16 of 36 patches examined. When TSN was added into the pipette solution, ion channels were observed in only 14 of 31 patches examined. Under both of these conditions, the probability that a toxin-induced channel would appear was reduced by approximately 50%. Moreover, the conductance and activity of the channels that appeared were reduced by more than 50%. The reduction in conductance in part represented a decrease in the size of the toxin-induced pore. As we know, the size of the HC-formed pore is an essential factor for BoNT translocation. It seems likely that the LC is unable to pass through the reduced pore formed in the TSN-treated cells because of its large dimensions. Therefore, the LC of the toxin can not approach its substrate protein in the TSN-treated cells, a fact that has already been demonstrated in our previous study[18].
During the binding, internalization and translocation of the toxin, interaction and recognition between toxin and biomembrane must be involved. In a previous study, we demonstrated that TSN inhibited delayed rectifier K+ channels, irrespective of intracellular application or extracellular addition[20]. TSN can modulate the channels on both sides of the membrane, meaning that it is membrane permeant. Moreover, TSN can modulate various kinds of K+ channels[24–26] and L-type Ca2+ channels [27], whose common denominator appears to be that they are imbedded into, and span the membrane’s bilayer. Therefore we hypothesized that TSN might derange the membrane environment after entering the membrane bilayer, and hence prevent BoNT from interacting with the membrane. Similarly, the change in the bilayer microenvironment caused by TSN affects ion channel activity, and results in inhibition of K+ currents and an increase of Ca2+ influx.
Together, the data obtained in the present study indicate that the antibotulismic effect of TSN might be achieved by interfering with translocation of the toxin.
Acknowledgement
The authors acknowledge the help provided by Wen-ping WANG in carrying out the experiments.
References
- Simpson LL. Molecular pharmacology of botulinum toxin and tetanus toxin. Annu Rev Pharmacol Toxicol 1986;26:427-53.
- Swaminathan S, Eswaramoorthy S. Structural analysis of the catalytic and binding sites of Clostridium botulinum neurotoxin B. Nat Struct Biol 2000;7:693-9.
- Montecucco C, Papini E, Schiavo G. Bacterial protein toxins penetrate cells via a four-step mechanism. FEBS Lett 1994;346:92-8.
- Schiavo G, Matteoli M, Montecucco C. Neurotoxins affecting neuroexocytosis. Physiol Rev 2000;80:717-66.
- Sonawane ND, Thiagarajah JR, Verkman AS. Chloride concentration in endosomes measured using a ratioable fluorescent Cl– indicator: evidence for chloride accumulation during acidification. J Biol Chem 2002;277:5506-13.
- Hoch DH, Romero-Mira M, Ehrlich BE, Finkelstein A, DasGupta BR, Simpson LL, et al. Channels formed by botulinum, tetanus, and diphtheria toxins in planar lipid bilayers: relevance to translocation of proteins across membranes. Proc Natl Acad Sci USA 1985;82:1692-6.
- Donovan JJ, Middlebrook JL. Ion-conducting channels produced by botulinum toxin in planar lipid membranes. Biochemistry 1986;25:2872-6.
- Blaustein RO, Germann WJ, Finkelstein A, DasGupta BR. The N-terminal half of the heavy chain of botulinum type A neurotoxin forms channels in planar phospholipid bilayers. FEBS Lett 1987;226:115-20.
- Schmid A, Benz R, Just I, Aktories K. Interaction of Clostridium botulinum C2 toxin with lipid bilayer membranes. Formation of cation-selective channels and inhibition of channel function by chloroquine. J Biol Chem 1994;269:16706-11.
- Sheridan RE. Gating and permeability of ion channels produced by botulinum toxin types A and E in PC12 cell membranes. Toxicon 1998;36:703-17.
- Koriazova LK, Montal M. Translocation of botulinum neurotoxin light chain protease through the heavy chain channel. Nat Struct Biol 2003;10:13-8.
- Chang CC, Hsie TH, Chen SF, Liang HT. The structure of chuanliansu. Acta Chem Sin 1975;33:35-47.
- Shu GX, Liang XT. A correction of the structure of chuanliansu. Acta Chem Sin 1980;38:196-8.
- Wang YB, Wen YX. A comprehensive report on clinical ascaris anthelmintic therapeutic effect of toosendanin pills. J Tradit Chin Med 1959;262:46-9.
- Li PZ, Zhou J, Miao WY, Ding FH, Meng JY, Ye HJ, et al. Therapeutic effect of toosendanin on animal botulism. Tradit Herb Drugs 1982;13:28-30. Chinese..
- Shi YL, Wang ZF. Cure of experimental botulism and antibotuli-smic effect of toosendanin. Acta Pharmacol Sin 2004;25:839-48.
- Shi YL, Xu K. Antibotulismic effect of toosendanin. Chin Sci Bull 1983;28:885-7.
- Zhou JY, Wang ZF, Ren XM, Tang MZ, Shi YL. Antagonism of botulinum toxin type A-induced cleavage of SNAP-25 in rat cerebral synaptosome by toosendanin. FEBS Lett 2003;555:375-9.
- Ray P, Berman JD, Middleton W, Brendle J. Botulinum toxin inhibits arachidonic acid release associated with acetylcholine release from PC12 cells. J Biol Chem 1993;268:11057-64.
- Hu Q, Huang FS, Shi YL. Inhibition of toosendanin on the delayed rectifier potassium current in neuroblastoma×glioma NG108-15 cells. Brain Res 1997;751:47-53.
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch 1981;391:85-100.
- Simpson LL. Ammonium chloride and methylamine hydrochloride antagonize clostridial neurotoxins. J Pharmacol Exp Ther 1983;225:546-52.
- Simpson LL, Coffield JA, Bakry N. Inhibition of vacuolar adenosine triphosphatase antagonizes the effects of clostridial neurotoxins but not phospholipase A2 neurotoxins. J Pharmacol Exp Ther 1994;269:256-62.
- Wang ZF, Shi YL. Inhibition of large-conductance Ca2+-activated K+ channels in hippocampal neurons by toosendanin. Neuroscience 2001;104:41-7.
- Wang ZF, Shi YL. Toosendanin-induced inhibition of small-conductance calcium-activated potassium channels in CA1 pyramidal neurons of rat hippocampus. Neurosci Lett 2001;303:13-6.
- Wang ZF, Shi YL. Modulation of inward rectifier potassium channel by toosendanin, a presynaptic blocker. Neurosci Res 2001;40:211-5.
- Li MF, Shi YL. The long-term effect of toosendanin on current through nifedipine-sensitive Ca2+ channels in NG108-15 cells. Toxicon 2004;45:53-60.