
Role of melatonin in Alzheimer-like neurodegeneration1
Introduction
Melatonin (N-acetyl-5-methoxytryptamine), a tryptophan metabolite, is synthesized mainly by the pineal gland. Melatonin has a number of physiological functions, including regulating circadian rhythms, clearing free radicals, improving immunity, and generally inhibiting the oxidation of biomolecules. It is generally accepted that melatonin deficit is closely related to aging and age-related diseases[1]. Decreased levels of melatonin in serum and in cerebrospinal fluid (CSF) and the loss of melatonin diurnal rhythm are observed in patients with Alzheimer disease (AD)[2−6]. Interestingly, the level of melatonin in CSF decreases with the progression of AD neuropathology as determined by the Braak stages[6]. Melatonin levels both in CSF and in postmortem human pineal gland are already reduced in preclinical AD subjects, who are cognitively still intact and have only the earliest signs of AD neuropathology[2,6]. A strong correlation exists between pineal content and CSF level of melatonin[2] and between CSF and plasma melatonin levels[7], suggesting that a reduced CSF melatonin level may serve as an early marker for the very first stages of AD. Although the pineal gland of AD patients has molecular changes, no changes in pineal weight, calcification or total protein content have been observed[2,8]. A recent study showed that β1-adrenergic receptor mRNA disappeared, and MAOA activity and gene expression were upregulated in AD patients, suggesting that the dysregulation of noradrenergic innervations and the depletion of serotonin, the precursor of melatonin, might be responsible for the loss of melatonin rhythm and reduced melatonin levels in AD[2]. In AD patients, melatonin supplementation has been suggested to improve circadian rhythmicity, for example decreasing agitated behavior, confusion and 搒undowning?, and to produce beneficial effects on memory in AD[9-13]. Therefore, melatonin supplementation may be one of the possible strategies for symptomatic treatment. Moreover, melatonin treatment seems to be safe because of its marked low toxicity[14,15]. However, adverse drug reactions may occur, such as (i) fever on the first day of melatonin treatment, which is possibly a reaction to the thermoregulatory function of melatonin; (ii) hyperkinesia or complaints of restless legs; (iii) menorrhagia, which may be explained by a decrease in plasma follicle-stimulating hormone (FSH) and luteinizing hormone (LH); (iv) pigmentation on arms and legs; (v) headache and abdominal reactions, such as nausea, dyspepsia and abdominal pain; (vi) thrombosis; and (vii) drowsiness[16,17]. When a pharmacological dose of melatonin (3.0 mg) is administered to elderly people, it not only induces sleep but also induces hypothermia. Moreover, intravenous administration of melatonin to schizophrenic patients in remission causes a worsening of psychotic symptoms, which persists even after the treatment is interrupted[18]. Pregnant women should avoid melatonin, because its (functional) teratological effects are not known. Additionally, there are also concerns with regard to the potential vasoactive nature of melatonin.
There are two characteristic pathologies in the brains of patients with AD: neurofibrillary tangles (NFT) composed of hyperphosphorylated microtubule-associated protein tau, and senile plaques (SP) mainly composed of Aβ peptide, derivations from the proteolytic processing of amyloid precursor protein (APP)[19]. Convincing evidence indicates that Aβ can mediate neurotoxicity through a complex series of interactions that involves increasing free radicals, raising intracellular calcium concentrations, and even triggering apoptosis[20]. Aβ is generally believed to play a major role in promoting neuronal degeneration by rendering neurons more vulnerable to age-related increases in levels of oxidative stress and impairments in cellular energy metabolism[21]. Tau protein is a major microtubule-associated protein that promotes microtubule assembly and stabilizes microtubules; it also takes part in the formation and maintenance of the axonal structure[22]. Hyperphosphorylated tau reduces the ability to stabilize microtubules, leading to disruption of the cytoskeletal arrangement and neuronal transport[23,24]. The extent of neurofibrillary pathology, and particularly the number of cortical NFT, correlates positively with the severity of dementia[25]. Although the exact pathogenesis of AD is not fully understood, both Aβ deposition and tau hyperphos-phorylation play critical roles in the development of AD. Because melatonin is able to improve some of the clinical symptoms of AD, and because melatonin levels decrease dramatically during AD, studies on the relationship between melatonin and tau/Aβ pathology will be helpful to understand and to assess its potential in the prevention or treatment of AD. This review will address the protective effects of melatonin on the pathological development of AD. Most of the data presented here are from animal studies, because the efficacy of melatonin in preclinical AD is currently not well documented.
Role of melatonin in tau hyperphosphorylation
Effect of melatonin on cytoskeletal reorganization The cytoskeleton plays a key role in maintaining the highly asymmetrical shape and structural polarity of neurons that are essential for neuronal physiology, and cytoskeletal reorganization plays a key role in neurogenesis. In neurodegenerative diseases, the cytoskeleton is abnormally assembled, and impairment of neurotransmission occurs[26,27]. Current data indicate that melatonin promotes neurogenesis through cytoskeletal rearrangements in a receptor-dependent and possibly subtype-selective manner[24,25]. Melatonin receptor 1 (MT1) is thought to be responsible for melatonin-induced neurite formation[28,29]. In addition, to promote microtubule rearrangement through Ca2+/calmodulin antagonism[30], melatonin could modulate phosphorylation and organization of vimentin intermediate filaments via protein kinase C activation in N1E-115 cells[31,32]. Alterations in cytoskeletal organization and melatonin levels in aging and neurodegenerative diseases support the notion that cyto-skeletal disruption is presumably associated with melatonin deficiency. These observations prompted us to investigate whether melatonin has beneficial effects on tau hyperphosphorylation, one of the characteristic pathological features in the AD brain.
Inhibition of tau hyperphosphorylation by melatonin Cytoskeletal alterations in AD are predominantly characterized by intracellular NFT mainly composed of an abnormal hyperphosphorylated form of the microtubule-associated protein, tau[33]. In healthy neurons, tau binds and stabilizes microtubules, which make up the cytoskeleton of the cell, by a reversible enzymatically mediated phosphorylation and dephosphorylation process. Hyperphosphorylation of tau leads to a decreased affinity with microtubules and the disruption of the neuronal cytoskeleton, as well as resulting in resistance to proteolytic degradation and gradual accumulation in the cell body[34].
The phosphorylation of tau is strictly regulated by a panel of protein phosphatases and protein kinases[35]. Inhibition of tau hyperphosphorylation is one target in AD treatment. Recently, we systemically studied the effect of melatonin on tau hyperphosphorylation induced by a series of activators of protein kinases and inhibitors of protein phosphatases. We found that melatonin efficiently attenuates tau or neurofilament hyperphosphorylation induced by wortmannin[36], calyculin A[37,38] and okadaic acid[39] in N2a and SH-SY5Y neuroblastoma cells. Our in vivo studies further demonstrated that melatonin significantly ameliorated tau hyper-phosphorylation elicited by wortmannin[40] and isoprotere-nol[41,42] in rats. To elucidate the mechanisms underlying the inhibitory effect of melatonin on tau hyperphos-phorylation, alterations of the activities of protein kinases and phosphatases were detected. Melatonin treatment not only inhibited wortmannin-induced glycogen synthase kinase-3 (GSK-3) activation, isoproterenol-induced protein kinase A (PKA) activation, and calyculin A-induced protein phosphatase-2A (PP-2A) inactivation, but also antagonized the oxidative stress induced by these agents [34,35,40]. These results from our studies provide supportive evidence for the strong efficacy of melatonin supplementation in inhibiting tau hyperphosphorylation induced by other stimuli. The next question is whether a decrease in melatonin levels would alter the phosphorylation state of the tau protein. To answer this question, we inhibited melatonin biosynthesis by injecting haloperidol, an inhibitor of 5-hydroxyindole-O-methyltransferase (one of the key enzymes in melatonin synthesis), into the lateral ventricle and the peritoneal cavity in rats[43]. The decreased serum level of melatonin, as detected by fluorescence high-performance liquid chromato-graphy, confirmed the successful inhibition of melatonin biosynthesis by haloperidol. More importantly, we found that inhibition of melatonin biosynthesis not only resulted in spatial memory impairment in rats, but also induced a reduction in tau phosphorylation with a concomitant decrease in PP-2A activity. Supplementation with melatonin by prior injection for 1 week and reinforcement during the haloperidol administration period significantly improved memory retention deficits, arrested tau hyperphosphory-lation and oxidative stress, and restored PP-2A activity[43]. As far as we know, this is the first report providing direct evidence for the physiological regulation by melatonin of tau phosphorylation, and PP-2A activity, as well as spatial memory. This finding is of great interest and significance because of the profoundly decreased melatonin levels and reduction in PP-2A activity in AD brain[44]. Although it is unclear whether diminished melatonin concentration is one of the causative factors or only a secondary process in AD pathology is unclear, our results strongly implicate decreased melatonin in Alzheimer-like spatial memory impairment and tau hyperphosphoryla-tion, as well as reduced PP-2A activity. Melatonin may play an important role in maintaining the physiological activity of PP-2A through a currently unknown mechanism, and decreased melatonin may be critical in the development of neurofibrillary degeneration. As haloperidol not only inhibits 5-hydroxyindole-O-methyltransferase, but also antagonizes dopamine D2 receptors, it is important to explore more selective means to suppress melatonin levels in the brains of experimental animals.
Possible mechanisms underlying the effect of melatonin on tau hyperphosphorylation Chemical agents used in our studies, including wortmannin, isoproterenol and calyculin A, not only induced tau phosphorylation, but also initiated oxidative stress, as manifested by an elevated level of malondialdehyde and an altered activity of superoxide dismutase[36,37,39]. Furthermore, melatonin is a potent direct free radical scavenger and indirect antioxidant that acts by augmenting the activity of several important antioxidative enzymes, for example superoxide dismutase, glutathione peroxidase and glutathione reductase[45]. Oxidative stress is known to influence the phosphorylation state of tau[46−48]. In a more recent study we have also demonstrated that calyculin A, a selective inhibitor of protein phosphatase of PP-2A and PP-1 that has little or no direct effect on other phosphatases or kinases, induced a significant activation of GSK-3 via oxidative stress[35]. It is therefore possible that prevention against tau phosphorylation by melatonin is partially due to antioxidant activity.
However, more importantly, although the precise underlying mechanisms are not fully understood, melatonin may act as an enzyme modulator in a way that is unrelated to its antioxidant properties. Accumulating data provide evidence for the regulation by melatonin of protein kinases including PKA[49,50], protein kinase C (PKC)[29,51], Ca2+/calmodulin-dependent kinase II(CaMKII)[52], and the mitogen-activated protein kinase (MAPK) family[53]. The documented correlation between melatonin and cAMP indicates that melatonin might inhibit PKA activity through the melatonin receptor-coupled inhibition of adenylyl cyclase and reduction of cAMP[49,50]. Although there is no evidence of a direct relationship between melatonin and GSK-3 activity, a recent study has found that melatonin treatment leads to elevated phosphorylation of Akt[54], an upstream regulator of GSK-3. It is possible that melatonin might at least partially inhibit GSK-3 activity through activating the phosphatidyl inositol-3 kinase (PI-3K)/Akt signaling pathway. Considering the regulation of GSK-3 activity by other protein kinases, we cannot exclude the possible contribution of melatonin-induced activation of the PKC and MAPK families. Based on our own studies and those by other groups, we believe that melatonin may function as an upstream modulator of extensive protein kinases and protein phosphatases, and GSK-3 is one of the most implicated as targets (Figure 1). Further study is necessary to fully elucidate the signal transduction modulated by melatonin.
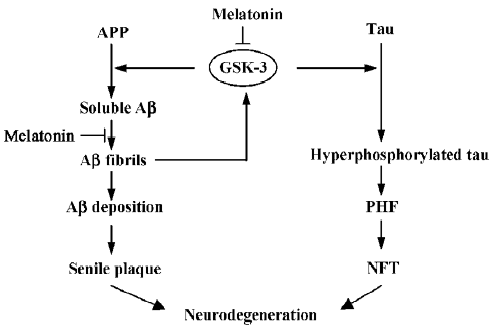
Melatonin and Aβ toxicity
Regulation by melatonin of Aβ generation Aβ is composed of 39−43 amino acid residues derived from proteolytic processing of a large precursor, APP, and plays a pivotal role in the dysfunction and death of neurons in AD[19,21]. Mature APP is processed proteolytically by distinct α-secretase or β-secretase pathways[55]. The nonamyloido-genic α-pathway involves the cleavage of APP within the Aβ sequence by α-secretase to release an N-terminal APP fragment, which in turn is cleaved by γ-secretase. Thus, the cleavage by γ-secretase precludes the formation of Aβ. The amyloidogenic β-secretase pathway, which results in the formation of intact Aβ peptide, is mediated by the sequential cleavage of β-secretase and γ-secretases at the N- and C-terminals of the Aβ sequence, respectively[64].
Melatonin has been found to have regulatory effects on APP metabolism. Melatonin treatment inhibited normal levels of secretion of soluble APP (sAPP) in different cell lines by interfering with APP full maturation[56]. Melatonin also affected the mRNA level of APP in a cell type-specific manner. Pretreatment with melatonin resulted in a significant reduction in the APP mRNA level in PC12 cells, but failed to produce this effect in human neuroblastoma cells[57]. We have also demonstrated that melatonin reduces Aβ generation in mouse neuroblastoma N2a cells harboring APP695[58]. An in vivo study showed that melatonin did not affect the expression of APP holoprotein in transgenic Tg2576 mice[59]. Addi-tionally, administration of melatonin efficiently reduced Aβ generation and deposition in vivo[59,60] and in vitro[56-58,61]. However, a recent study showed that, despite achieving high plasma concentrations of melatonin, chronic melatonin therapy in old Tg2576 mice initiated at 14 months of age not only failed to remove existing plaques, but also failed to prevent additional Aβ deposition. This result is in contrast with those of diminished Aβ in melatonin-treated wild type mice[60] and reduced Aβ and protein nitration in melatonin-treated Tg2576 mice[59]. The age at initiation of melatonin treatment may be the key difference that accounts for the discrepancy between the studies of Matsubara et al[59] and Quinn et al[62], in which the same transgenic Tg2576 mouse model was used. Amyloid plaque pathology typically appears in Tg2576 mice at 10‒12 months of age[63]. Melatonin treatment in the study of Matsubara et al was started at 4 months of age (prior to the appearance of hippocampal and cortical plaques)[59], an earlier pathological stage compared with 14 months of age in the study of Quinn et al[62]. However, both studies concur in finding little evidence of the potent antioxidant effects of melatonin in the oldest mice. These findings indicate that melatonin has the ability to regulate APP metabolism and prevent Aβ pathology, but fails to exert anti-amyloid or antioxidant effects when initiated after the age of Aβ deposition.
Although consistent conclusions were achieved, none of the related studies further explain how melatonin exerts its inhibitory effect on Aβ generation. The proteolytic cleavage of APP by the α-secretase pathway is regulated by many physiological and pathological stimuli; the PKC-dependent mechanism is one of the most recognized. Stimuli such as muscarinic and metabotrophic glutamate receptor agonists and phorbol esters share the capacity to stimulate soluble APP secretion and inhibit Aβ formation through PKC activation[64]. The mechanism whereby PKC activity increases soluble APP secretion is still unknown, but it may involve additional kinase steps and the eventual activation of the secretases that mediated APP cleavage. Recently, the inhibitory regulation by GSK-3 on Aβ generation has been well established[65-67]. GSK-3 interacts with presenilin-1, a cofactor for γ-secretase, implying that GSK-3 may function as a component in the γ-secretase complex[68,69]. Assuming that melatonin can influence PKC and GSK-3 activity as mentioned earlier, it is postulated that melatonin may regulate APP processing through the PKC and GSK-3 pathways. Because PKC is an upstream regulator of GSK-3, GSK-3 may be one of the common signal pathways that regulate both Aβ generation and tau hyperphosphorylation (Figure 1).
Regulation by melatonin of the formation of Aβ fibrils Important pathological properties of Aβ, such as neurotoxicity and resistance to proteolytic degradation, depend on the ability of peptides to form β-sheet structures and/or amyloid fibrils[70,71]. Intervention in the Aβ aggregation process can be considered an approach to stopping or slowing the progression of AD. Melatonin can interact with Aβ40 and Aβ42 and inhibit the progressive formation of β-sheet and/or amyloid fibrils[72-74]. The antifibrillogenic effect of melatonin has been demonstrated by different techniques, including circular dichroism (CD) spectroscopy, electron microscopy and nuclear magnetic resonance (NMR) spectroscopy, and electrospray ionization mass spectrometry (ESI-MS)[73]. Moreover, the interaction between melatonin and Aβ appears to depend on the structural characteristics of melatonin rather than on its antioxidant properties, because it could not be reproduced by melatonin analogs or other free radical scavengers[70,72]. Evidence derived from ESI-MS proved that there was a hydrophobic interaction between Aβ and melatonin, and proteolytic investigations suggested that the interaction took place on the 29‒40 residues of the Aβ segment[73]. Results from NMR spectroscopy further confirmed a residue-specific interaction between melatonin and any of the three histidine and aspartate residues of Aβ[74]. The imidazole-carboxylate salt bridges formed by the side chains of histidine (His+) and aspartate (Asp?) residues are critical to the formation of the amyloid β-sheet structures[75], and disruption of these salt bridges promotes fibril dissolution[76]. Melatonin could promote the conversion of β-sheets into random coils by disrupting the imidazole-carboxylate salt bridges and thus prevent Aβ fibrillogenesis and aggregation. It is therefore possible that by blocking the formation of the secondary β-sheet conformation, melatonin may not only reduce neurotoxicity but also facilitate clearance of the peptide via increased proteolytic degradation.
Protective effects of melatonin on Aβ-induced toxicity It has been postulated that Aβ, a major component of SP, is responsible for the neuronal degeneration observed in the vicinity of SP in the AD brain, and is responsible for the disease pathology. Aβ treatment elicits a spectrum of cellular damage, including increases in lipid peroxidation and intracellular free calcium concentration, oxidative damage to mitochondrial DNA, and the emergence of apoptotic markers[18]. Oxidative stress acts synergistically with disturbance of intracellular calcium homeostasis: the free radical-induced membrane damage induces further calcium influx, and the resultant accentuated calcium influx in turn will induce the generation of further free radicals. Therefore, oxidative stress plays a central role in Aβ-induced neurotoxicity, and even cell death. Aside from Aβ causing oxidative stress, it has been proposed that oxidative damage could exacerbate a vicious cycle, in which amyloidogenic processing of APP would be further facilitated to generate more Aβ, which in turn enhances oxidative stress[77]. Accumulating data implies that melatonin efficiently protects cells against Aβ-induced oxidative damage and cell death in vitro[78,79] and in vivo [59,80-82]. In Aβ-treated cells and animals, melatonin exerts its protective activity mainly through an antioxidant effect, whereas in APP-transfected cells and transgenic animal models, the underlying mechanism is attributed to not only its antioxidant property, but also its anti-amyloid property, including inhibition of both Aβ generation and formation of β-sheets and/or amyloid fibrils. However, it is difficult to determine the extent of the contribution from each of these properties to the overall effects of melatonin treatment in vivo.
Other neuroprotective effects of melatonin
Protection of the cholinergic system In cholinergic neurons, the synthesis of ACh by choline acetyltransferase (ChAT) is dependent on intracellular pools of choline provided by high-affinity choline uptake and of acetyl-CoA replenished by metabolism. In a subsequent step, newly synthesized ACh is transported into synaptic vesicles by the energy-dependent vesicular ACh transporter. Biological investigations of tissue from biopsy and autopsy have found a profound decrease in the activity of the ACh-synthesis enzyme, ChAT, in the neocortex, which correlates positively with the severity of dementia[83]. Reduced choline uptake, ACh release and loss of cholinergic neurons from the basal forebrain region further indicate a selective presynaptic cholinergic deficit in the hippocampus and neocortex of individuals with AD. The degeneration of the cholinergic innervation from the basal forebrain to the hippocampal formation in the temporal lobe is thought to be one of the factors determining the progression of memory decay both during normal aging and AD. The mechanism of reduced cholinergic function remains unclear, but it is thought that Aβ has negative effects on multiple aspects of ACh synthesis and release, including choline uptake, ChAT activity and ACh release[83]. A previous study has also demonstrated that melatonin partially prevented peroxynitrite-induced inhibition of choline transport and ChAT activity[84]. Recently, Feng et al reported that 8-month-old APP695 transgenic mice had not only Aβ deposition but also significant learning and memory deficits, as well as cholinergic system dysfunction, as indicated by a profound reduction in ChAT activity in the frontal cortex and hippocampus. Long-term melatonin treatment (4 months) significantly ameliorated such neuropatho-logical, behavioral and biochemical changes in APP695 transgenic mice[80]. Another study by Feng by et al also showed that similar treatment with melatonin antagonized spatial memory impairment and decreased ChAT activity in ovariectomized adult rats[85]. Additionally, an inhibitory effect of melatonin on ACh release was found in intact rabbit retinas[86]. These findings indicate that protection of the cholinergic system may occur at multiple steps that are critical for ACh synthesis and release (Figure 2).
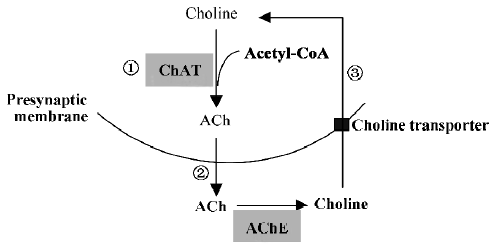
Anti-inflammatory actions Epidemiological studies have shown that non-steroidal anti-inflammatory drug (NSAID) use decreases the incidence of AD[87]. Aβ itself has been shown to act as a proinflammatory agent that causes the activation of many inflammatory components, and SP co-exist with cytokines and chemokines, and are surrounded by microglia and astrocytes, indicating the involvement of inflammation in the pathogenesis of AD[88]. The activated microglia induced by Aβ are the major sources of inflammatory response. Microglial activation might involve beta-amyloid binding and the activation of cell surface immune and adhesion molecules[89]. It has been reported that melatonin attenuates kainic acid-induced microglial and astroglial responses as determined by immunohistochemical detection of isolectin-B4 and glial fibrillary acidic protein (GFAP), the specific markers for microglia and astroglia, respectively[90]. Oral melatonin administration also attenuated Aβ-induced proinflammatory cytokines in rat brain[82]. A recent study showed the potent suppressive effect of melatonin pretreatment on the ischemia-reperfusion-stimulated ipsilateral increase in the immunoreactivity for neuronal NO synthase (nNOS), cyclooxyrenase-2 (COX-2) or myeloperoxidase (MPO), but not for GFAP, suggesting the contribution of anti-inflammatory response in neuroprotection against ischemia-reperfusion damage[91]. Nuclear factor-kappaB (NF-κB) plays an important role in inflammatory mediator-mediated signal transduction. Melatonin has been shown to inhibit tumor necrosis factor (TNF) and brain injury-induced NF-κB activation[92,93]. These results suggest that the anti-inflammatory properties of melatonin are due to inhibition of the production of inflammatory mediators and downstream signal transduction.
Expectations
Melatonin has been proposed as a treatment for AD based on the fact that the level of melatonin reduces during aging and in AD patients, as well as for its antioxidant and anti-amyloid effects. Recent studies from APP transgenic mice have indicated that early, long-term melatonin supplementation produces anti-amyloid and antioxidant effects, but no such effect is produced when melatonin treatment is initiated after the age of amyloid formation[58-61]. Extensive clinical trials and studies with transgenic models are necessary to confirm the role of melatonin at the late pathological stage of AD. If melatonin has no effect at the late stage of AD, studies on melatonin should be limited to the prevention of AD, rather than treatment. Our studies have demonstrated the efficacy of melatonin in the inhibition of tau hyperphos-phorylation. As mentioned earlier, GSK-3 may be one of the common signaling pathways that regulates both Aβ generation and tau phosphorylation. Moreover, GSK-3 is considered to be a key protein kinase involved in Aβ-induced tau hyperphosphorylation. Therefore, the regulation by melatonin of protein kinases, especially GSK-3, is worthy of further exploration. Although GSK-3 has been implicated as a central player in the altered metabolism of both tau and Aβ, mechanistic links between melatonin and GSK-3 have so far been only indirectly evidenced by the sole fact that phosphorylation of a GSK-3 regulator, Akt, in the brain is augmented by treatment with melatonin. Future research strategies will need to overcome the current limitations on insights into the direct functional coupling between melatonin and GSK-3. For instance, experimental paradigms such as the one provided by a recent study using a mouse model of tauopathy[94] could be applied to examination of the hypothesis of melatonin- and GSK-3-mediated neurodegen-erative tau pathogenesis. In mammals melatonin exerts some of its functions through two specific high-affinity membrane receptors belonging to the superfamily of G-protein-coupled receptors: MT1 and MT2. Decreased MT2 immunoreactivity and increased MT1 immunoreactivity have been reported in the hippocampus of AD patients[95,96]. The connection between changes in receptor expression and the unsatisfactory therapeutic effects of melatonin when started at a late pathological stage of AD remain to be elucidated.
As a conclusion, in addition to its well-established antioxidant effect, melatonin prevents cells from Aβ-mediated toxicity not only by inhibiting Aβ generation but also by inhibiting Aβ aggregation and formation of amyloid fibrils. Furthermore, melatonin attenuates tau hyperphosphorylation induced by activation of protein kinases or inhibition of protein phosphatases. Moreover, melatonin may be involved in the physilogical regulation of tau phosphorylation. Early, long-term application of melatonin may at least slow down the development of AD.
References
- Wu YH, Swaab DF. The human pineal gland and melatonin in aging and Alzheimer抯 disease. J Pineal Res 2005;38:145-52.
- Wu YH, Feenstra MG, Zhou JN, Liu RY, Torano JS, van Kan HJ, et al. Molecular changes underlying reduced pineal melatonin levels in Alzheimer disease: alterations in preclinical and clinical stages. J Clin Endocrinol Metab 2003;88:5898-906.
- Ferrari E, Arcaini A, Gornati R, Pelanconi L, Cravello L, Fioravanti M, et al. Pineal and pituitary-adrenocortical function in physiological aging and in senile dementia. Exp Gerontol 2000;35:1239-50.
- Ohashi Y, Okamoto N, Uchida K, Iyo M, Mori N, Morita Y. Daily rhythm of serum melatonin levels and effect of light exposure in patients with dementia of the Alzheimer抯 type. Biol Psychiatry 1999;45:1646-52.
- Liu RY, Zhou JN, van Heerikhuize J, Hofman MA, Swaab DF. Decreased melatonin levels in postmortem cerebrospinal fluid in relation to aging, Alzheimer抯 disease, and apolipoprotein E-epsilon4/4 genotype. J Clin Endocrinol Metab 1999;84:323-7.
- Zhou JN, Liu RY, Kamphorst W, Hofman MA, Swaab DF. Early neuropathological Alzheimer抯 changes in aged individuals are accompanied by decreased cerebrospinal fluid melatonin levels. J Pineal Res 2003;35:125-30.
- Rousseau A, Petren S, Plannthin J, Eklundh T, Nordin C. Serum and cerebrospinal fluid concentrations of melatonin: a pilot study in healthy male volunteers. J Neural Transm 1999;106:883-8.
- Friedland RP, Luxenberg JS, Koss E. A quantitative study of intracranial calcification in dementia of the Alzheimer type. Int Psychogeriatr 1990;2:36-43.
- Cohen-Mansfield J, Garfinkel D, Lipson S. Melatonin for treatment of sundowning in elderly persons with dementia: a preliminary study. Arch Gerontol Geriatr 2000;31:65-76.
- Brusco LI, Marquez M, Cardinali DP. Melatonin treatment stabilizes chronobiologic and cognitive symptoms in Alzheimer抯 disease. Neuro Endocrinol Lett 2000;21:39-42.
- Brusco LI, Marquez M, Cardinali DP. Monozygotic twins with Alzheimer抯 disease treated with melatonin: case report. J Pineal Res 1998;25:260-63.
- Cardinali DP, Brusco LI, Lloret SP, Furio AM. Melatonin in sleep disorders and jet-lag. Neuro Endocrinol Lett 2002;23 Suppl 1:9-13.
- Cardinali DP, Brusco LI, Liberczuk C, Furio AM. The use of melatonin in Alzheimer抯 disease. Neuro Endocrinol Lett 2002;23 Suppl 1:20-2.
- Karasek M, Reiter RJ, Cardinali DP, Pawlikowski M. Future of melatonin as a therapeutic agent. Neuroendocrinol Lett 2002;23 Suppl 1:118-21.
- Singer C, Tractenberg RE, Kaye J, Schafer K, Gamst A, Grundman M, et al. Alzheimer抯 disease cooperative study. A multicenter, placebo-controlled trial of melatonin for sleep disturbance in Alzheimer抯 disease. Sleep 2003;26:893-90.
- Nagtegaal J, Kerkhof GA, Smits MG, van der Meer YG, Fischer-Steenvoorden MGJ. Melatonin: a survey of suspected adverse drug reactions. In: Beersma DGM, Coenen AML, editors. Sleep-wake research in the Netherlands, vol 7; 1996. p 115‒8.
- Avery D, Lenz M, Landis C. Guidelines for prescribing melatonin. Ann Med 1998;30:122-30.
- Pacchierottio C, Iapichino S, Bossini L, Pieraccini F, Castrogiovanni P. Melatonin in psychiatric disorders: a review on the melatonin involvement in psychiatry. Front Neuroendocrinol 2001;22:18-32.
- Selkoe DJ. Alzheimer抯 disease is a synaptic failure. Science 2002;298:789-91.
- Eckert A, Keil U, Marques CA, Bonert A, Frey C, Schussel K, et al. Mitochondrial dysfunction, apoptotic cell death, and Alzheimer抯 disease. Biochem Pharmacol 2003;66:1627-34.
- Selkoe DJ. Cell biology of protein misfolding: the examples of Alzheimer抯 and Parkinson抯 diseases. Nat Cell Biol 2004;6:1054-61.
- Saragoni L, Hernandez P, Maccioni RB. Differential association of tau with subsets of microtubules containing posttranslationally-modified tubulin variants in neuroblastoma cells. Neurochem Res 2000;25:59-70.
- Brion JP, Anderton BH, Authelet M, Dayanandan R, Leroy K, Lovestone S, et al. Neurofibrillary tangles and tau phosphory-lation. Biochem Soc Symp 2001;67:81-8.
- Billingsley ML, Kincaid RL. Regulated phosphorylation and dephosphorylation of tau protein: effects on microtubule interaction, intracellular trafficking and neurodegeneration. Biochem J 1997;323:577-91.
- Braak E, Braak H, Mandelkow EM. A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol (Berl) 1994;87:554-67.
- Grundke-Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem 1986;261:6084-9.
- Lee VM, Balin BJ, Otvos L Jr, Trojanowski JQ. A68: a major subunit of paired helical filaments and derivatized forms of normal Tau. Science 1991;251:675-8.
- Bordt SL, McKeon RM, Li PK, Witt-Enderby PA, Melan MA. N1E-115 mouse neuroblastoma cells express MT1 melatonin receptors and produce neurites in response to melatonin. Biochim Biophys Acta 2001;1499:257-64.
- Witt-Enderby PA, MacKenzie RS, McKeon RM, Carroll EA, Bordt SL, Melan MA. Melatonin induction of filamentous structures in non-neuronal cells that is dependent on expression of the human mt1 melatonin receptor. Cell Motil Cytoskeleton 2000;46:28-42.
- Benitez-King G, Tunez I, Bellon A, Ortiz GG, Anton-Tay F. Melatonin prevents cytoskeletal alterations and oxidative stress induced by okadaic acid in N1E-115 cells. Exp Neurol 2003;182:151-9.
- Benitez-King G. PKC activation by melatonin modulates vimentin intermediate filament organization in N1E-115 cells. J Pineal Res 2000;29:8-14.
- Benitez-King G, Hernandez ME, Tovar R, Ramirez G. Melatonin activates PKC-alpha but not PKC-epsilon in N1E-115 cells. Neurochem Int 2001;39:95-102.
- Eidenmuller J, Fath T, Hellwig A, Reed J, Sontag E, Brandt R. Structural and functional implications of tau hyperphosphoryla-tion: information from phosphorylation-mimicking mutated tau proteins. Biochemistry 2000;39:13166-75.
- Avila J. Tau aggregation into fibrillar polymers: taupathies. FEBS Lett 2000;476:82-92.
- Gong CX, Liu F, Grundke-Iqbal I, Iqbal K. Post-translational modifications of tau protein in Alzheimer抯 disease. J Neural Transm 2005;112:813-38.
- Deng YQ, Xu GG, Duan P, Zhang Q, Wang JZ. Effects of melatonin on wortmannin-induced tau hyperphosphorylation. Acta Pharmacol Sin 2005;26:519-26.
- Li XC, Wang ZF, Zhang JX, Wang Q, Wang JZ. Effect of melatonin on calyculin A-induced tau hyperphosphorylation. Eur J Pharmacol 2005;510:25-30.
- Li SP, Deng YQ, Wang XC, Wang YP, Wang JZ. Melatonin protects SH-SY5Y neuroblastoma cells from calyculin A-induced neurofilament impairment and neurotoxicity. J Pineal Res 2004;36:186-191.
- Wang YP, Li XT, Liu SJ, Zhou XW, Wang XC, Wang JZ. Melatonin ameliorated okadaic-acid induced Alzheimer-like lesions. Acta Pharmacol Sin 2004;25:276-80.
- Liu SJ, Wang JZ. Alzheimer-like tau phosphorylation induced by wortmannin in vivo and its attenuation by melatonin. Acta Pharmacol Sin 2002;23:183-7.
- Wang DL, Ling ZQ, Cao FY, Zhu LQ, Wang JZ. Melatonin attenuates isoproterenol-induced protein kinase A overactivation and tau hyperphosphorylation in rat brain. J Pineal Res 2004;37:11-16.
- Wang XC, Zhang J, Yu X, Han L, Zhou ZT, Zhang Y, et al. Prevention of isoproterenol-induced tau hyperphosphorylation by melatonin in the rat. Acta Physiol Sin 2005;57:7-12.
- Zhu LQ, Wang SH, Ling ZQ, Wang DL, Wang JZ. Effect of inhibiting melatonin biosynthesis on spatial memory retention and tau phosphorylation in rat. J Pineal Res 2004;37:71-7.
- Gong CX, Shaikh S, Wang JZ, Zaidi T, Grundke-Iqbal I, Iqbal K. Phosphatase activity toward abnormally phosphorylated tau: decrease in Alzheimer disease brain. J Neurochem 1995;65:732-8.
- Reiter RJ, Acuna-Castroviejo D, Tan DX, Burkhardt S. Free radical-mediated molecular damage: mechanisms for the protective actions of melatonin in the central nervous system. Ann NY Acad Sci 2001;939:200-15.
- Zhu X, Rottkamp CA, Boux H, Takeda A, Perry G, Smith MA. Activation of p38 kinase links tau phosphorylation, oxidative stress, and cell cycle-related events in Alzheimer disease. J Neuropathol Exp Neurol 2000;59:880-8.
- Gomez-Ramos A, Diaz-Nido J, Smith MA, Perry G, Avila J. Effect of the lipid peroxidation product acrolein on tau phosphorylation in neural cells. J Neurosci Res 2003;71:863-70.
- Lovell MA, Xiong S, Xie C, Davies P, Markesbery WR. Induction of hyperphosphorylated tau in primary rat cortical neuron cultures mediated by oxidative stress and glycogen synthase kinase-3. J Alzheimers Dis 2004;6:659-71.
- Schuster C, Williams LM, Morris A, Morgan PJ, Barrett P. The human MT1 melatonin receptor stimulates cAMP production in the human neuroblastoma cell line SH-SY5Y cells via a calcium-calmodulin signal transduction pathway. J Neuroendocrinol 2005;17:170-8.
- Peschke E, Muhlbauer E, Musshoff U, Csernus VJ, Chankiewitz E, Peschke D. Receptor (MT(1)) mediated influence of melatonin on cAMP concentration and insulin secretion of rat insulinoma cells INS-1. J Pineal Res 2002;33:63-71.
- Rivera-Bermudez MA, Gerdin MJ, Earnest DJ, Dubocovich ML. Regulation of basal rhythmicity in protein kinase C activity by melatonin in immortalized rat suprachiasmatic nucleus cells. Neurosci Lett 2003;346:37-40.
- Benitez-King G, Rios A, Martinez A, Anton-Tay F. In vitro inhibition of Ca2+/calmodulin-dependent kinase II activity by melatonin. Biochim Biophys Acta 1996;1290:191-6.
- Chan AS, Lai FP, Lo RK, Voyno-Yasenetskaya TA, Stanbridge EJ, Wong YH. Melatonin mt1 and MT2 receptors stimulate c-Jun N-terminal kinase via pertussis toxin-sensitive and -insensitive G proteins. Cell Signal 2002;14:249-57.
- Kilic U, Kilic E, Reiter RJ, Bassetti CL, Hermann DM. Signal transduction pathways involved in melatonin-induced neuroprotection after focal cerebral ischemia in mice. J Pineal Res 2005;38:67-71.
- Selkoe DJ. The cell biology of beta-amyloid precursor protein and presenilin in Alzheimer抯 disease. Trends Cell Biol 1998;8:447-53.
- Lahiri DK. Melatonin affects the metabolism of the beta-amyloid precursor protein in different cell types. J Pineal Res 1999;26:137-46.
- Song W, Lahiri DK. Melatonin alters the metabolism of the beta-amyloid precursor protein in the neuroendocrine cell line PC12. J Mol Neurosci 1997;9:75-92.
- Zhang YC, Wang ZF, Wang Q, Wang YP, Wang JZ. Melatonin attenuates overproduction of β-amyloid-induced inhibition in expression of neurofilament protein. Acta Pharmacol Sin 2004;25:447-51.
- Matsubara E, Bryant-Thomas T, Pacheco Quinto J, Henry TL, Poeggeler B, Herbert D, et al. Melatonin increases survival and inhibits oxidative and amyloid pathology in a transgenic model of Alzheimer抯 disease. J Neurochem 2003;85:1101-8.
- Lahiri DK, Chen D, Ge YW, Bondy SC, Sharman EH. Dietary supplementation with melatonin reduces levels of amyloid beta-peptides in the murine cerebral cortex. J Pineal Res 2004;36:224-31.
- Olivieri G, Hess C, Savaskan E, Ly C, Meier F, Baysang G, et al. Melatonin protects SHSY5Y neuroblastoma cells from cobalt-induced oxidative stress, neurotoxicity and increased beta-amyloid secretion. J Pineal Res 2001;31:320-5.
- Quinn J, Kulhanek D, Nowlin J, Jones R, Pratico D, Rokach J, et al. Chronic melatonin therapy fails to alter amyloid burden or oxidative damage in old Tg2576 mice: implications for clinical trials. Brain Res 2005;1037:209-13.
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, et al. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 1996;274:99-102.
- Fisher A, Pittel Z, Haring R, Bar-Ner N, Kliger-Spatz M, Natan N, et al. M1 muscarinic agonists can modulate some of the hallmarks in Alzheimer抯 disease: implications in future therapy. J Mol Neurosci 2003;20:349-56.
- Su Y, Ryder J, Li B, Wu X, Fox N, Solenberg P, et al. Lithium, a common drug for bipolar disorder treatment, regulates amyloid-beta precursor protein processing. Biochemistry 2004;43:6899-908.
- Ryder J, Su Y, Liu F, Li B, Zhou Y, Ni B. Divergent roles of GSK3 and CDK5 in APP processing. Biochem Biophys Res Commun 2003;312:922-9.
- Phiel CJ, Wilson CA, Lee VM, Klein PS. GSK-3alpha regulates production of Alzheimer抯 disease amyloid-beta peptides. Nature 2003;423:435-9.
- Tesco G, Tanzi RE. GSK3 beta forms a tetrameric complex with endogenous PS1-CTF/NTF and beta-catenin. Effects of the D257/D385A and FAD-linked mutations. Ann NY Acad Sci 2000;920:227-32.
- Takashima A, Murayama M, Murayama O, Kohno T, Honda T, Yasutake K, et al. Presenilin 1 associates with glycogen synthase kinase-3beta and its substrate tau. Proc Natl Acad Sci USA 1998;95:9637-41.
- Simmons LK, May PC, Tomaselli KJ, Rydel RE, Fuson KS, Brigham EF, et al. Secondary structure of amyloid beta peptide correlates with neurotoxic activity in vivo. Mol Pharmacol 1994;45:373-9.
- Soto C, Castano EM. The conformation of Alzheimer抯 beta peptide determines the rate of amyloid formation and its resistance to proteolysis. Biochem J 1996;314:701-7.
- Poeggeler B, Miravalle L, Zagorski MG, Wisniewski T, Chyan YJ, Zhang Y, et al. Melatonin reverses the profibrillogenic activity of apolipoprotein E4 on the Alzheimer amyloid Abeta peptide. Biochemistry 2001;40:14995-5001.
- Skribanek Z, Balaspiri L, Mak M. Interaction between synthetic amyloid-peptide (1-40) and its aggregation inhibitors studied by electrospray ionization mass spectrometry. J Mass Spectrom 2001;36:1226-9.
- Pappolla M, Bozner P, Soto C, Shao H, Robakisi NK, Zagorski M, et al. Inhibition of Alzheimer β-fibrillogenesis by melatonin. J Biol Chem 1998;273:7185-8.
- Huang TH, Fraser PE, Chakrabartty A. Fibrillogenesis of Alzheimer Abeta peptides studied by fluorescence energy transfer. J Mol Biol 1997;269:214-24.
- Fraser PE, Nguyen JT, Surewicz WK, Kirschner DA. pH-dependent structural transitions of Alzheimer amyloid peptides. Biophys J 1991;60:1190-201.
- Bieschke J, Zhang Q, Powers ET, Lerner RA, Kelly JW. Oxidative metabolites accelerate Alzheimer抯 amyloidogenesis by a two-step mechanism, eliminating the requirement for nucleation. Biochemistry 2005;44:4977-83.
- Feng Z, Zhang JT. Protective effect of melatonin on beta-amyloid-induced apoptosis in rat astroglioma C6 cells and its mechanism. Free Radic Biol Med 2004;37:1790-801.
- Zatta P, Tognon G, Carampin P. Melatonin prevents free radical formation due to the interaction between beta-amyloid peptides and metal ions J Pineal Res 2003;35:98-103. [Al(III), Zn(II), Cu(II), Mn(II), Fe(II)].
- Feng Z, Chang Y, Cheng Y, Zhang BL, Qu ZW, Qin C, et al. Melatonin alleviates behavioral deficits associated with apoptosis and cholinergic system dysfunction in the APP 695 transgenic mouse model of Alzheimer抯 disease. J Pineal Res 2004;37:129-36.
- Shen YX, Xu SY, Wei W, Sun XX, Liu LH, Yang J, et al. The pro-tective effects of melatonin from oxidative damage induced by amyloid beta-peptide 25-35 in middle-aged rats. J Pineal Res 2002;32:85-9.
- Rosales-Corral S, Tan DX, Reiter RJ, Valdivia-Velazquez M, Martinez-Barboza G, Acosta-Martinez JP, et al. Orally administered melatonin reduces oxidative stress and proinflamma-tory cytokines induced by amyloid-beta peptide in rat brain: a comparative, in vivo study versus vitamin C and E. J Pineal Res 2003;35:80-4.
- Kar S, Slowikowski SP, Westaway D, Mount HT. Interaction between β-amyloid and central cholinergic neurons: implications for Alzheimer抯 disease. J Psychiatry Neurosci 2004;29:427-41.
- Guermonprez L, Ducrocq C, Gaudry-Talarmain YM. Inhibition of acetylcholine synthesis and tyrosine nitration induced by peroxynitrite are differentially prevented by antioxidants. Mol Pharmacol 2001;60:838-46.
- Feng Z, Cheng Y, Zhang JT. Long-term effects of melatonin or 17 beta-estradiol on improving spatial memory performance in cognitively impaired, ovariectomized adult rats. J Pineal Res 2004;37:198-206.
- Mitchell CK, Redburn DA. Melatonin inhibits ACh release from rabbit retina. Vis Neurosci 1991;7:479-86.
- Stuchbury G, Munch G. Alzheimer抯 associated inflammation, potential drug targets and future therapies. J Neural Transm 2005;112:429-53.
- Tuppo EE, Arias HR. The role of inflammation in Alzheimer抯 disease. Int J Biochem Cell Biol 2005;37:289-305.
- Ho GJ, Drego R, Hakimian E, Masliah E. Mechanisms of cell signaling and inflammation in Alzheimer抯 disease. Curr Drug Targets Inflamm Allergy 2005;4:247-56.
- Chung SY, Han SH. Melatonin attenuates kainic acid-induced hippocampal neurodegeneration and oxidative stress through microglial inhibition. J Pineal Res 2003;34:95-102.
- Pei Z, Cheung RT. Pretreatment with melatonin exerts anti-inflammatory effects against ischemia/reperfusion injury in a rat middle cerebral artery occlusion stroke model. J Pineal Res 2004;37:85-91.
- Beni SM, Kohen R, Reiter RJ, Tan DX, Shohami E. Melatonin-induced neuroprotection after closed head injury is associated with increased brain antioxidants and attenuated late-phase activation of NF-kappaB and AP-1. FASEB J 2004;18:149-151.
- Sasaki M, Jordan P, Joh T, Itoh M, Jenkins M, Pavlick K, et al. Melatonin reduces TNF-α induced expression of MAdCAM-1 via inhibition of NF-kappaB. BMC Gastroenterol 2002;2:9.
- Noble W, Planel E, Zehr C, Olm V, Meyerson J, Suleman F, et al. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc Natl Acad Sci USA 2005;102:6990-5.
- Savaskan E, Olivieri G, Meier F, Brydon L, Jockers R, Ravid R, et al. Increased melatonin 1α-receptor immunoreactivity in the hippocampus of Alzheimer抯 disease patients. J Pineal Res 2002;32:59-62.
- Savaskan E, Ayoub MA, Ravid R, Angeloni D, Fraschini F, Meier F, et al. Reduced hippocampal MT2 melatonin receptor expression in Alzheimer抯 disease. J Pineal Res 2005;38:10-6.